INTRODUCTION
The outbreak of coronavirus disease-19 (COVID-19), caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), affected more than 777 million people worldwide (1). Multiple Variants of Concern (VOCs) and Variants of Interest (VOIs) of SARS-CoV-2 emerged with increased transmissibility, severity, and ability to evade the host immune response (2). The continued incidence and emergence of novel variants necessitate the development of advanced diagnostic testing methods for COVID-19.
The main diagnostic tools for SARS-CoV-2 infection are nucleic acid amplification and immunodiagnostic tests (3). The diagnostic pipeline for polymerase chain reaction (PCR)- based methods involves isolating total RNA from nasopharyngeal or oropharyngeal swab specimens and amplifying the existing viral genome using primers specific to the viral genes. The earliest COVID-19 diagnostic test was performed using reverse transcription polymerase chain reaction (RT-PCR). Reverse transcription loop-mediated isothermal amplification (RT-LAMP) is an efficient method with a rapid turnaround time (4, 5, 6). Immunodiagnostic tests detect the presence of viral proteins or host antibodies in samples from patients with COVID-19. Serological detection methods for SARS-CoV-2 include enzyme-linked immunosorbent assays, lateral flow immunoassays, and luminescent immunoassays (7, 8, 9). Laboratory-based diagnostic tests require skilled personnel and heavy instrumentation and are inaccessible in remote settings (10). It is crucial to widely deploy inexpensive, sensitive, and specific diagnostic platforms to stop the spread of COVID-19.
Point-of-care (POC) molecular diagnostic tests in non-laboratory settings have enabled the rapid detection and identification of SARS-CoV-2 (11). Representative POC tests such as Abbott ID NOW COVID-19, Xpert Xpress SARS-CoV-2, and AcculaSARS-CoV-2 Test have been approved for emergency use (12). However, these existing detection modules have low throughput, which restricts their use in large-scale testing in a short interval (13, 14). Furthermore, the mutations in the N2 region of the delta variant reduce the sensitivity of Xpert Xpress SARS-CoV-2 (15). Conventional LAMP assays can produce false-positives due to non-specific amplification (16). Of all the variants of SARS-CoV-2 that have been analyzed, Omicron has the highest number of mutation sites associated with increased binding affinity, immune evasion and transmissibility. The rapid mutation rate and emergence of new SARS-CoV-2 variants require ongoing updates to diagnostic tests to ensure the detection of multiple SARS-CoV-2 targets while maintaining insensitivity to genetic variations (17). A portable real time PCR diagnostic platform, the Biomeme Franklin™ Real-Time PCR thermocycler, enables the detection of multiple targets in a single reaction and the visualization of the test result on a smartphone application. The device enables detection of 27 targets in a sample or testing of a total of nine samples in a single run. Additionally, this device is a battery-powered and affordable POC diagnostic tool in low- and mid-income settings to accurately track infections (18). The portable Biomeme RT-qPCR assay has been confirmed as a POC test for the detection of canine distemper and Ebola viruses (19, 20).
This study aimed at developing and validating one-step multiplex detection test of SARS-CoV-2 using the Biomeme Franklin™ thermocycler. For this testing method, we compared the efficiency of various commercial RNA extraction kits using the convenient TRIzol approach. We evaluated the Biomeme assay against a benchtop RT-qPCR platform, QuantStudio 3, for the detection of SARS-CoV-2. The assay targets conserved regions in nucleocapsid, envelope and spike genes of the SARS-CoV-2 to minimize false-negative results due to genetic variability of the virus. We demonstrated the utility of this one-step multiplex RT-qPCR test for SARS-CoV-2 detection using nasopharyngeal specimens from patients in the Republic of Korea (ROK).
MATERIAL AND METHODS
Ethical states
In-vitro infection with SARS-CoV-2 was performed at Biosafety Level 3 at the Hallym Clinical and Translational Science Institute at Hallym University in Chuncheon, Republic of Korea. The study was conducted following the institutional biosafety requirements and guidelines outlined in Hallym2020-04 on Oct. 30, 2020, as approved by the Hallym University Institutional Biosafety Committee. The infected patient residual specimens were collected from 28th February 2022 to 7th March 2023 and collection was approved by the Institutional Review Board (IRB) of Hallym University (HIRB-2021-091) (Supplementary Table 1). The ethics committee waived patient consent. The specimens were transported in viral transport medium [AB Transport Medium (ABTM) & Swabs, AB Medical, Seoul, Republic of Korea].
Cell line, viruses, and in-vitro infection
The African green monkey kidney epithelial cells Vero E6 (ATCC® CRL-1596) were cultivated in Dulbecco Modified Eagle Medium (Cat No. 11995065, Gibco®, Life technologies, Europe B.V) supplemented with 10% Fetal Bovine Serum (FBS; Cat No. 10082147, Gibco®, Life technologies, Europe B.V), 1% of 10 mM HEPES in 0.85% NaCl (Cat No. 17-737E, Lonza, BioWhittaker®, Walkersville, MD, USA), and 1% antibiotic-antimycotic (Cat No. 15240062, Gibco®, Life technologies, Europe B.V). Cells were maintained at 37 °C with 5% CO2. Severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2 19A (NCCP No. 43326), B.1.617.2, (NCCP No. 43406), BA.1, (NCCP No. 43408), BA.2 (NCCP No. 43412), BA.2.75 (NCCP No. 43417), BA.5 (NCCP No. 43426), and BN.1 (NCCP No. 43439)] were acquired from the National Culture Collection for Pathogens (Osong, ROK).
A total of 1×106 Vero E6 cells were infected at multiplicity of infection (MOI) of 0.01 for 2 h, rotating plate every 20 min. At forty-eight hours post-infection (hpi), the cells were harvested.
RNA extraction and cDNA synthesis
RNA extraction from infected cell samples was performed using TRIzol (AMBION Inc., Austin, TX, USA) according to manufacturer’s protocol and commercial kits. Cell culture-derived virus was first inactivated in the lysis buffer contained in the M1 Sample Prep Cartridge Kit (Cat No. 3000536R, Biomeme Inc., Philadelphia, PA, USA). Briefly, 500 µL of virus-infected cell samples were lysed by mixing in 1mL of Biomeme Lysis Buffer (BLB), which was then passed through the M1 sample cleanup column using the supplied 1 mL lock syringe and washed. Finally, nucleic acid was eluted in the elution buffer (100 µL) provided. For employment of the QIAamp Viral Mini Kit (Cat No. 52904, Qiagen, Hilden, Germany), infected cells were first lysed, and the sample was loaded onto the QIAamp Mini spin column and washed. Subsequently RNA was eluted from the column using 60 µL of nuclease free water. For employment of XpressAmp™ Direct Amplification Reagents (Cat No. A8882, Promega, Wisconsin, USA), harvested cell samples were mixed with lysis buffer and 1-thioglycerol to reach a concentration of 1% (v/v). After pipetting and incubation at 25 °C for 10 min, 5 µL of the prepared sample lysates were used for subsequent cDNA synthesis. cDNA synthesis was performed using a High-Capacity RNA-to-cDNA kit (Cat No. 4387406, Applied Biosystems, Foster City, CA, USA).
Reverse transcription-quantitative polymerase chain reaction
RT-qPCR was conducted using SYBR Green PCR Master mix (Applied Biosystems) and cycling conditions including denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 65 °C for 1 min on a QuantStudio 3 Real-Time PCR Instrument (A28132, Applied Biosystems, Foster City, CA, USA) and Franklin® Real-Time PCR Thermocycler (1000003, Biomeme Inc., Philadelphia, PA, USA; Fig. 1).

Fig. 1
Workflow of Biomeme assay. 1. The Biomeme Franklin is set up using the power button on the device. 2. The RNA extracted is combined with primers, probes and multiplex master mixture in the empty Go-Strips placed on a rack. 3. The tubes are loaded into holes in the device. 4. The lid of the thermocycler is closed, and the reaction starts, and the reaction settings and results are operated using the smartphone application. 5. The result is reported by the smartphone.
Primers targeting viral nucleocapsid (N) and envelope (E) genes were used according to the guidelines of a previous study (21). To design spike (S) gene specific primers and probes, a total of seven strains of SARS-CoV-2 including 19A, B.1.617.2, BA.1, BA.2, BA.2.75, BA.5, and BN.1 were aligned using the Clustal W method with the Lasergene program, version 5 (DNASTAR). S gene specific primers and probes were designed in the conserved region. The primer and probe sequences are listed in Supplementary Table 2. The primers were analyzed in-silico for the detection of self- and cross-dimer using ThermoFisher Multiple Primer analyzer tool. The relative gene copy number was calculated using GAPDH as the internal control.
Sensitivity, specificity, and qPCR efficiency calculations
The total RNA recovered from cells infected with SARS-CoV-2 19 A and variants (B.1.617.2, BA.1, BA.2, BA.2.75, BA.5, and BN.1) was diluted to the same concentration of 1000 ng/uL. Then, four 100-fold serial dilutions were prepared in nuclease free water. Sensitivity was determined using four serial 100-fold dilutions of total RNA to generate a standard curve using SYBR Green PCR Master mix. The dilutions were tested in triplicates. PCR efficiency was calculated using the equation: E = [10 (-1/slope)-1) x 100]. For specificity testing, RNA (100 ng) derived from virus infected cells was used to synthesize cDNA. Subsequently, the cycle threshold (Ct) values were obtained for triplicate wells on QuantStudio 3 and Biomeme.
One-step multiplex RT-qPCR for Biomeme assay
Two kits were compared for one-step multiplex RT-qPCR using the primers and probes listed in Supplementary Table 2 using QuantStudio 3 and Biomeme.
The TaqPath™ 1-step Multiplex Master Mix (Cat No. A28525, Applied Biosystem, Foster City, CA, USA) conditions included incubation step at 25 °C for 2 min; reverse transcription step at 53 °C for 10 min; polymerase activation step at 95 °C for 2 min, and 40 cycles of 95 °C for 3 s, and 60 °C for 30 s. The 20 µL reaction mixture contained 5 µL of TaqPath 1-step Multiplex Master Mix (4X), 1 μL of 5 µM primers, 0.5 µL of 5 µM probes, and 1 µL of RNA template from ten-fold serial dilutions.
The One Step PrimeScript III RT-qPCR Mix (Cat No. RR600A, Takara, Shiga, Japan) conditions included reverse transcription reaction at 25 °C for 10 min, 52 °C for 5 min and inactivation reaction at 95 °C for 10 s, followed by 40 cycles at 95 °C for 5 s, 60 °C for 30 s. The reaction mixture of 50 µL contained 10 µL of One Step PrimeScript III RT-qPCR Mix (2X), 0.8 µL of 5 µM primer mix, 0.8 μL of 5 µM probes and 1 μL of RNA template from 10-fold serial dilutions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (Version 10.2.1; GraphPad Software, Inc., La Jolla, CA). Values are presented in bar graphs as the mean ± SD (standard deviation) of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 were considered statistically significant.
RESULTS
Evaluation and validation of various RNA extraction kits from SARS-CoV-2-infected cells
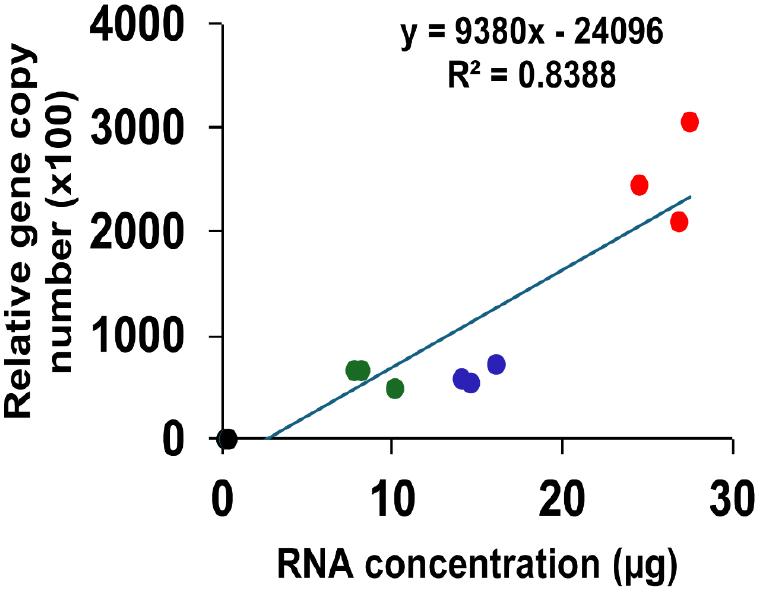
Total RNA was recovered from SARS-CoV-2 19A-infected Vero E6 cells using the TRIzol reagent and commercially available RNA purification kits. The highest amount of RNA was obtained using the TRIzol method (26.3 µg), followed by the QIAamp Viral RNA kit (13.0 µg), and M1 Sample Prep Cartridge Kit (10.0 µg). The lowest RNA yield was obtained with the XpressAmp™ Direct Amplification Reagents kit (0.3 µg; Fig. 2A). The obtained RNA samples were examined using RT-qPCR targeting the SARS-CoV-2 N gene to validate the RNA extraction kits used. The TRIzol-derived RNA samples exhibited the highest relative copy numbers of the N gene. The RNA samples obtained using the QIAamp Viral RNA kit and the M1 Sample Prep Cartridge Kit contained comparable relative copy numbers of the SARS-CoV-2 N gene. The lowest relative copy number of the SARS-CoV-2 N gene was noted in the RNA sample obtained using the XpressAmp™ Direct Amplification Reagents kit (Fig. 2B). The copy numbers of SARS-CoV-2 N gene correlated with the total amount of RNA extracted using the different kits evaluated in this study (R2 = 0.84, p = 2.882 × 10-5; Supplementary Fig. 1).
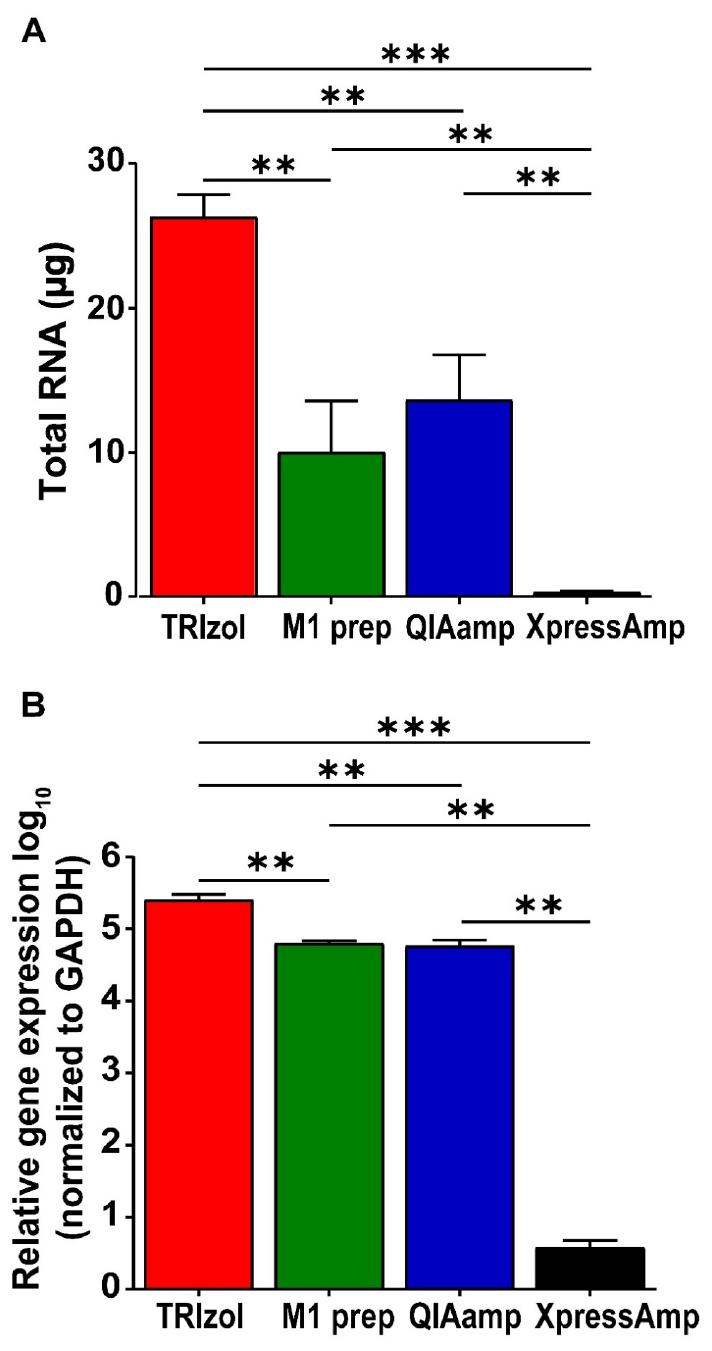
Fig. 2
Evaluation and comparison of RNA extraction kits for SARS-CoV-2 detection using RT-qPCR. Vero E6 cells were infected with SARS-CoV-2 19A for 48 h. Total cellular RNA was extracted using TRIzol reagent, the M1 sample prep cartridge kit, the QIAamp viral RNA kit, and XpressAmp™ Direct Amplification Reagents. Then, 100 ng of RNA was used for cDNA synthesis and analyzed using RT-qPCR on QuantStudio 3. (A) The total amount of RNA (μg) recovered from each method was measured using a NanoDrop spectrophotometer. (B) Data represent SARS-CoV-2 N gene expression levels normalized to GAPDH and are shown as relative expression values. The error bars represent the SD from three independent replicates. **p < 0.01, ***p < 0.001, one-way ANOVA.
Evaluation of sensitivity and specificity of the Biomeme assay for SARS-CoV-2 and variants
To estimate the sensitivity of the Biomeme assay, total RNA extracted from cells infected with the SARS-CoV-2 wild type and its variants were serially diluted and examined using RT-qPCR (Fig. 3). The average Ct values in the highest diluted total RNA samples ranged from 35.7 to 40.0 for the N, E, and S genes. SARS-CoV-2 RNA was detected at Ct values less than 32 from 0.01 ng/µl of total RNA using the Biomeme assay. The regression equations of the N, E, and S genes for the different SARS-CoV-2 variants analyzed using the Biomeme assay and QuantStudio 3 are shown in Supplementary Table 3.
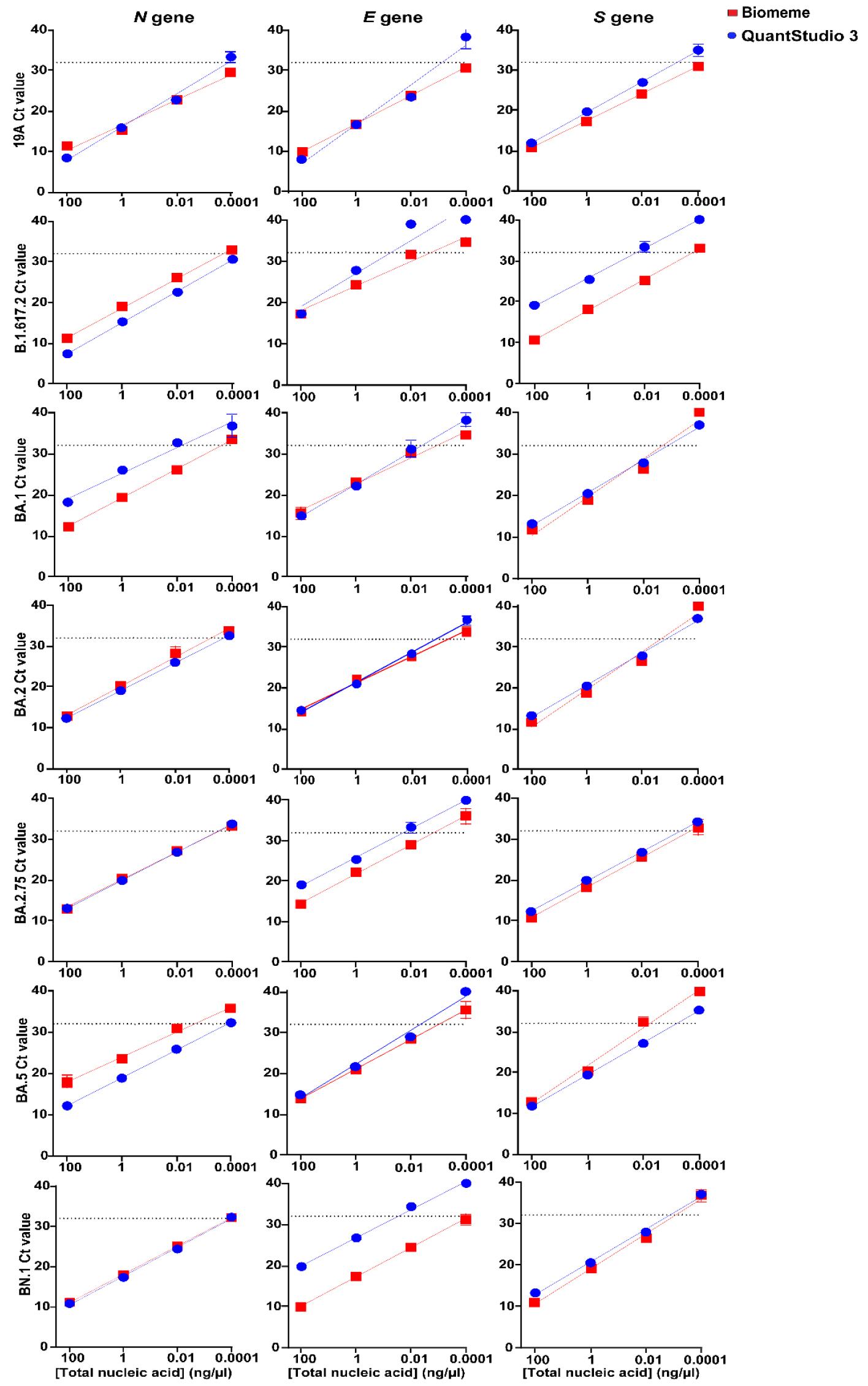
Fig. 3
Sensitivity of the Biomeme assay for SARS-CoV-2. The sensitivity of the Biomeme assay was determined using 100-fold serial dilutions of total RNA (from 100 ng/μl to 0.0001 ng/μl) extracted from cells infected with 19A and variants of SARS-CoV-2 at an MOI of 0.01. RT-qPCR was performed using SYBR-Green PCR mix and primers for three target genes. For each dilution, the Ct values on QuantStudio 3 and the Biomeme assay are plotted on the Y-axis. RNA amounts for the dilution series are labelled on the X-axis. The points represent the mean ± SD of triplicate Ct values obtained from each dilution. The dotted line represents the cutoff for the Biomeme assay corresponding to a Ct value of 32. Linear regression analysis was performed using GraphPad Prism.
We tested the specificity of the Biomeme assay for the SARS-CoV-2 wild type and variants and other common human respiratory viruses. The analysis of SARS-CoV-2 RNA samples on QuantStudio 3 resulted in Ct values ranging from 9.3 to 14.2 for the N gene, 9.1 to 21.0 for the E gene, and 11.8 to 19.1 for the S gene, whereas the corresponding values obtained with the Biomeme assay were 11.0 to 12.9, 10.0 to 17.3, and 10.7 to 12.9, respectively. All variants of SARS-CoV-2 included in the study were detected by both QuantStudio 3 and the Biomeme assay. Amplification for viruses other than SARS-CoV-2, including human coronavirus- OC43, -229E, and -NL63, was observed at Ct values above 32 for both methods (Fig. 4).
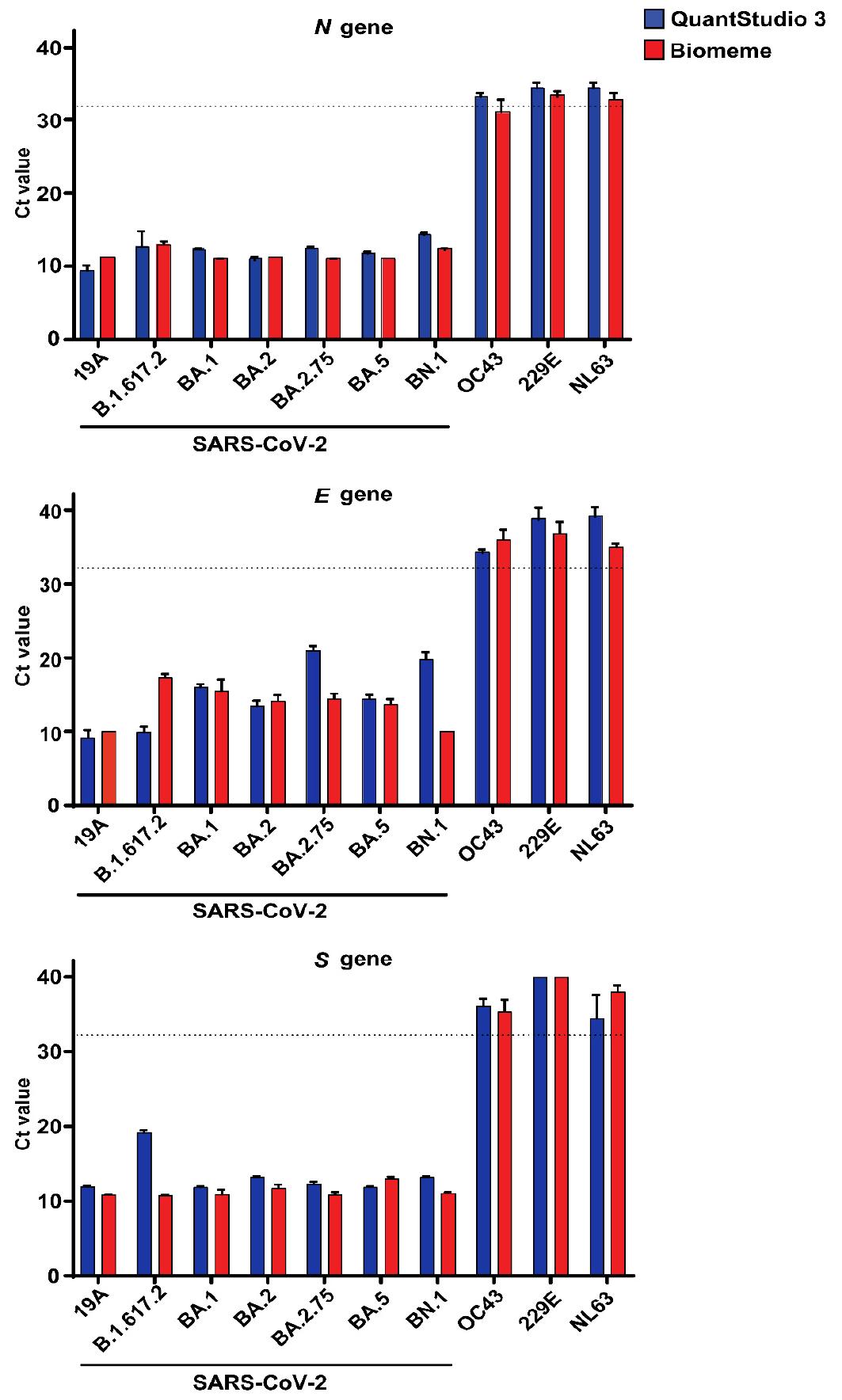
Fig. 4
Specificity of the Biomeme assay for SARS-CoV-2. The specificity of the Biomeme assay was determined using total RNA recovered from cells infected with wild type 19A and six different variants of SARS-CoV-2 and the human coronaviruses- OC43, NL63, and 229E at an MOI of 0.01 for 48 h. Then, 100 ng of total RNA was used for cDNA synthesis and Ct values were obtained from RT-qPCR on QuantStudio 3 and Biomeme using cDNA as template and SYBR Green for detection. The bars represent the mean ± SD of triplicate Ct values obtained via QuantStudio 3 and the Biomeme assay. The dotted line represents the cutoff for the Biomeme assay, corresponding to a Ct value of 32.
Evaluation of one-step multiplex RT-qPCR kits for the Biomeme-based SARS-CoV-2 assay
We evaluated two commercial one-step multiplex RT-qPCR reagent kits, TaqPath™ 1-Step Multiplex Master Mix and One Step PrimeScript III RT-qPCR Mix, to quantify SARS-CoV-2 19A RNA using the Biomeme platform. For all target genes, the mean Biomeme Ct values obtained using One Step PrimeScript III RT-qPCR Mix were two to three cycles higher than those from TaqPath™ 1-Step Multiplex Master Mix at the lowest RNA concentration (Fig. 5). The amplification efficiency for the N, E and S genes with One Step PrimeScript III RT-qPCR Mix was 63–71.0%. A qPCR amplification efficiency of 87.0–90.2% was obtained with TaqPath™ 1-Step Multiplex Master Mix using the Biomeme assay (Supplementary Table 4). Overall, TaqPath™ 1-Step Multiplex Master Mix showed high performance in the one-step multiplex RT-qPCR detection of SARS-CoV-2 RNA using the Biomeme assay.
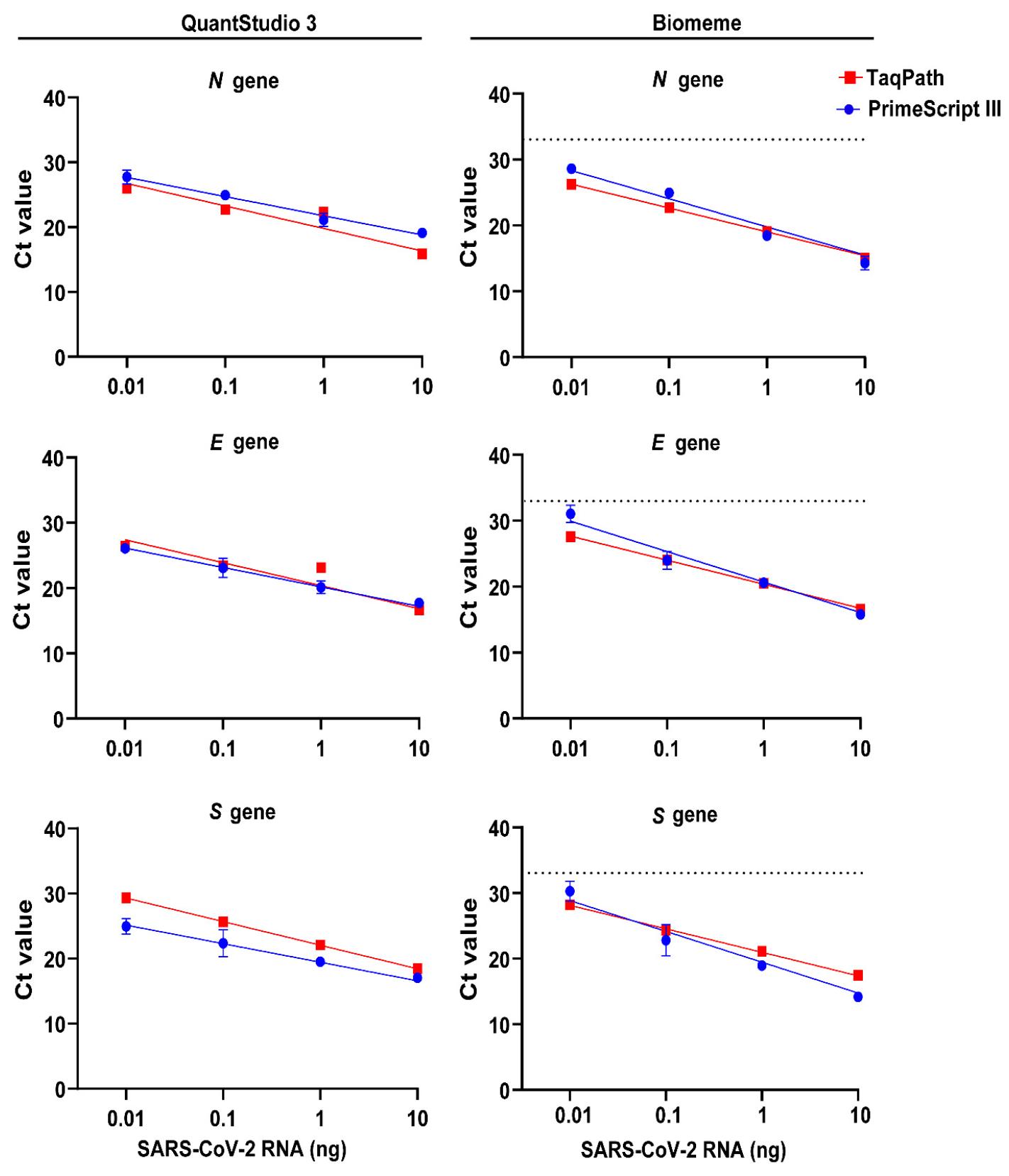
Fig. 5
Assessment of one-step RT-qPCR kits for SARS-CoV-2 RNA detection using the Biomeme platform. Total cellular RNA from SARS-CoV-2 19A-infected Vero E6 cells was extracted using TRIzol reagent. RNA was diluted in ten-fold serial dilutions and used as the template for one-step RT-qPCR. The comparison of Ct values from two commercial kits, Taqpath™ and PrimeScript III, for the Biomeme assay is shown for primers targeting the nucleocapsid (N), envelope (E) and spike (S) genes of SARS-CoV-2. Linear regression analysis was performed using GraphPad prism. Data represent the mean ± SD of triplicate Ct values. The dotted line represents the cutoff for the Biomeme assay, corresponding to a Ct value of 32.
Validation of the one-step multiplex RT-qPCR Biomeme assay for SARS-CoV-2 detection
We first tested primers and probes for the Biomeme assay for the detection of the viral genome obtained from cells infected with SARS-CoV-2 19A and variants in a singleplex format. The Ct values from the singleplex assay ranged from 10.7 to 16.0. To establish a one-step multiplex RT-qPCR assay for SARS-CoV-2 detection, we combined primers and probes specific to the N, E, and S genes of SARS-CoV-2. The multiplexed RT-PCR resulted in Biomeme Ct values between 10.6 and 17.8 and QuantStudio 3 Ct values between 11.3 and 17.3 (Fig. 6), detecting SARS-CoV-2 RNA from all variants and 19A. These results indicate that the Biomeme assay enables sensitive and consistent detection of a broad range of SARS-CoV-2 lineages under multiplexed RT-qPCR conditions.
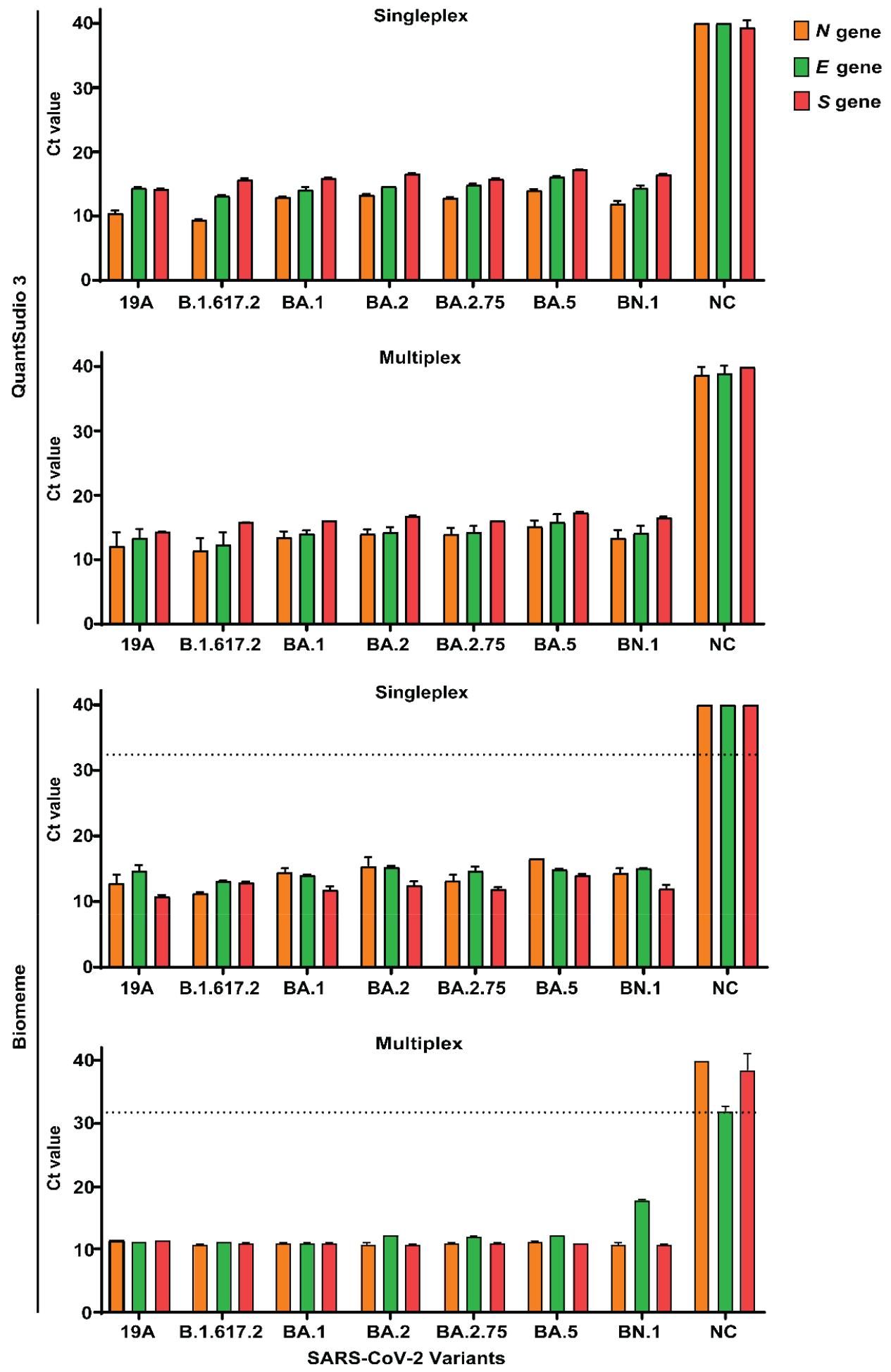
Fig. 6
Comparison of Ct values between singleplex and multiplex RT-qPCR formats for SARS-CoV-2 detection using the Biomeme thermocycler. TaqPath™ 1-Step Multiplex Master Mix was used for the RT-qPCR Biomeme assay on RNA derived from infected Vero E6 cells. (A) RT-qPCR measurements on QuantStudio 3, in which 100 ng of RNA template was added into reactions containing N, E, and S gene primer-probe mixtures in singleplex and multiplex formats. (B) Evaluation of Ct values from Biomeme thermocycler when 5 μM of N, E, and S gene primers and 5 μM probes are combined with 100 ng of RNA in total reaction volume of 20 μL. Data represent the mean ± SD of triplicate Ct values. The negative control (NC) was RNA from uninfected Vero E6 cells.
Analytical assessment of one-step multiplex RT-qPCR for SARS-CoV-2
To validate the performance of one-step multiplex RT-qPCR for POC detection of SARS-CoV-2, RNA from nasopharyngeal swabs of 40 (20 known cases of SARS-CoV-2 infection and 20 uninfected/healthy) patients in the ROK were tested using the Biomeme assay and QuantStudio 3 (Fig. 7). Based on the QuantStudio 3 results, 19 samples were identified as positive. The average Ct values in the RT-qPCR-positive cases ranged from 15.1 to 31.5 for the N gene, from 13.9 to 30.6 for the E gene, and from 13.8 to 30.3 for the S gene. RNA samples from the same patients were assessed using Biomeme, and 95% of the positive samples and 100% of the negative samples were correctly identified. The Ct values in Biomeme positive cases ranged from 12.7 to 31.4 for the N gene, from 13.4 to 30.7 for the E gene, and from 13.1 to 31.3 for the S gene. One indefinite positive sample showed Ct values in the range of 36 and 40 for all target genes using both Biomeme and QuantStudio 3. The overall sensitivity and specificity of Biomeme was 95 and 100%, respectively Supplementary Table 5. These findings demonstrate that Biomeme effectively detects SARS-CoV-2 RNA using a one-step multiplex RT-qPCR method targeting the N, E, and S genes.
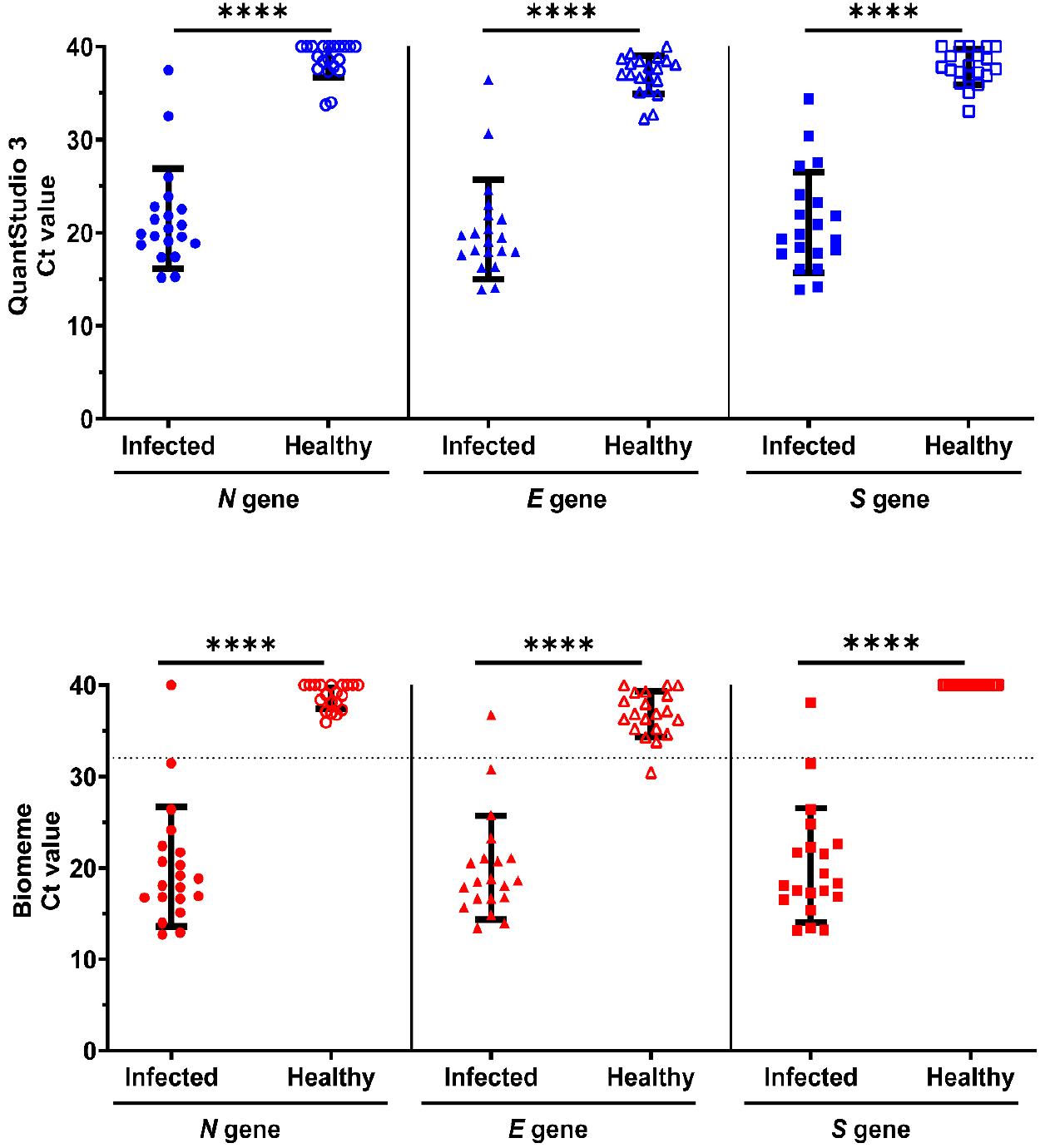
Fig. 7
Evaluation of Biomeme platform for one-step multiplex RT-qPCR detection of SARS-CoV-2 in nasopharyngeal swab samples. RNA from 40 nasopharyngeal swab samples was extracted using TRIzol LS reagent and analyzed using one-step multiplex RT-qPCR on the QuantStudio 3 and Biomeme platforms using 100 ng of RNA as the template. The Ct values from the RT-qPCR of SARS-CoV-2-infected and -uninfected/healthy samples using the N, E, and S primer–probe mixtures are indicated on the Y-axis. In the RT-qPCR on the Biomeme platform, one indefinite positive sample gave Ct values between 36 and 40 with the Biomeme assay. Each data point represents the mean Ct value of technical replicates for an individual clinical sample. Error bars represent the group mean ± SD. Statistical comparisons between infected and healthy groups were performed using an unpaired two-tailed t-test.****p < 0.0001.
DISCUSSION
In this study, we evaluated the suitability of the Biomeme Franklin thermocycler for the reliable testing of patients suspected to have SARS-CoV-2 infection. The proposed Biomeme platform sensitively and specifically detected the presence of the RNA of SARS-CoV-2 and its variants in infected cells. We performed one-step multiplex RT-qPCR to detect SARS-CoV-2 RNA in clinical samples to validate the clinical application, robustness, and sensitivity of the portable Biomeme thermocycler.
We tested the efficiency of commercial RNA purification kits in recovering sufficient amounts of RNA for the detection of SARS-CoV-2. Compared with the TRIzol RNA extraction method, the M1 Sample Prep cartridge kit showed a significantly lower yield of viral RNA. There was no difference in RNA yield between the silica membrane-based RNA extraction methods, including the M1 Sample Prep cartridge kit and the QIAamp Viral RNA Mini Kit (22). The performance of filtration-based RNA extraction methods could be influenced by multiple washing steps and membrane clogging due to incomplete lysis, thereby affecting downstream enzymatic reactions (23). Moreover, RNA quality is compromised without purification when using direct amplification reagents, potentially inhibiting PCR reaction. This study highlighted the sub-optimal performance of SARS-CoV-2 detection using the M1 Sample Prep cartridge kit as compared to the standard TRIzol method. Therefore, the RNA extraction kit for POC testing remains to be developed.
Our results showed that the RT-qPCR Biomeme assay could detect SARS-CoV-2 RNA in 0.01 ng/µl of total RNA with Ct values less than 32 using primers for the N, E, and S genes. We also demonstrated that the assay was robust in detecting multiple SARS-CoV-2 variants, an advantage not provided by antigen tests. Additionally, the molecular detection method utilized in the Biomeme assay can be modified to reduce gene drop-out linked with the emergence of new SARS-CoV-2 variants. Multiplex assays reduce processing time and optimize throughput, particularly in situations where consumables may be limited. A direct comparison of cost between the singleplex and multiplex detection remains to be investigated. Our comparative analysis of multiplex one-step RT-qPCR kits for the Biomeme assay showed higher amplification efficiency with TaqPath™ 1-Step Multiplex Master Mix than with One Step PrimeScript III RT-qPCR Mix for the same amount of RNA. Using TaqPath™ 1-Step Multiplex Master Mix with the Biomeme assay can thus minimize re-testing of inconclusive samples and save both cost and time in healthcare.
We found that the one-step multiplex RT-qPCR assay on the Biomeme thermocycler had high analytical sensitivity and specificity. We defined SARS-CoV-2 positivity as a Ct value less than 32. One clinical sample exhibited Ct values close to this threshold and was classified as positive, confirming the Biomeme assay’s ability to detect low viral loads. Inconclusively high Ct values were observed in one of the 20 infected samples across both the Biomeme and QuantStudio 3 platforms which may result from low quantities of viral RNA in samples collected in the early or end stages of infection, and follow-up testing is required in case of inconclusive results (24). Testing nasal swab samples using Biomeme SARS-CoV-2 Go-Strips demonstrated 98% analytical sensitivity and 100% specificity (25). Another study using ready-to-use Biomeme SARS-CoV-2 Go-Strips showed a sensitivity of 87% for detecting SARS-CoV-2 in clinical samples (26). These results are based on detection of SARS-CoV-2 gene targets open reading frame 1ab (ORF1ab) and spike (S). The performance of the Biomeme assay assessed in this study indicates accurate multiplex identification of viral RNA from viral targets including nucleocapsid, envelope and spike genes, and compatibility of the Biomeme assay with commercial one-step multiplex RT-qPCR kits. Using the Franklin thermocycler in low-resource settings can thus facilitate diagnostic testing.
For POC diagnosis, it is essential to reduce the turnaround time from sampling to the result, reduce hardware complexity, and ensure high sensitivity and specificity (27). Portable RT-PCR instruments with gold-standard accuracy can aid POC testing in remote settings. The Biomeme thermocycler is a compact and lightweight platform. It enables quantitative detection and sharing of results over smartphone devices or computers. The device can operate at 4-40 ℃ and 0-99% humidity (28). The miniaturized RT-qPCR-based Biomeme system validated in this study allows for rapid diagnosis in 3–4 h, all without the need for specialized infrastructure (Fig. 8). The simplicity of Biomeme thermocycler operation and its increased mobility are significant advantages for the deployment in community healthcare locations and hospital settings, including emergency departments and intensive care units. Given the benefits of multiplex PCR, the proposed Biomeme assay can be further developed for the detection of other respiratory pathogens or infectious diseases with specific target primer sets.
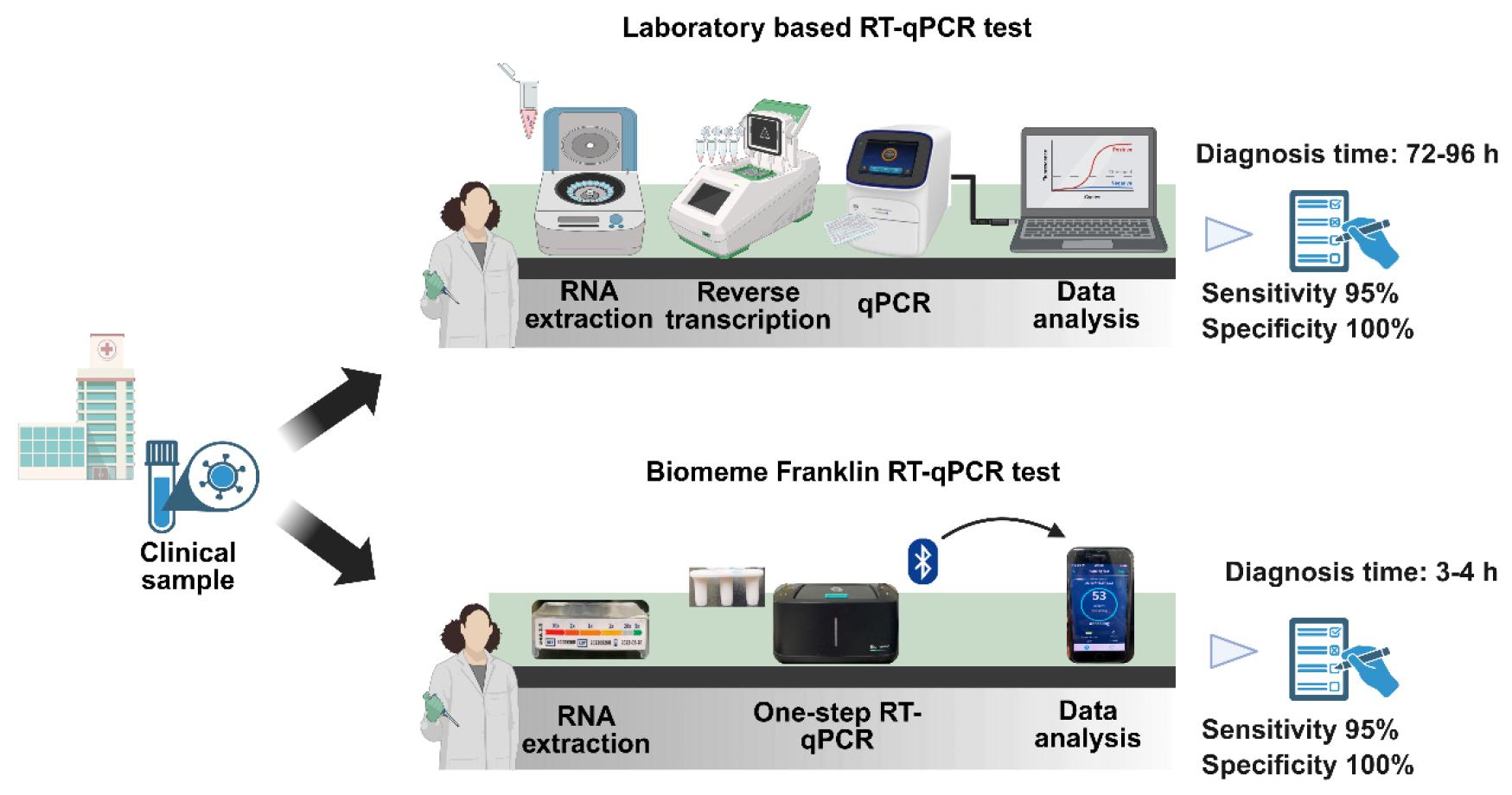
Fig. 8
Workflow for laboratory RT-qPCR and the Biomeme Franklin Thermocycler for detecting SARS-CoV-2 in clinical samples. For laboratory RT-qPCR diagnostic testing, the collected clinical sample is transported to a laboratory or near-patient setting. In the laboratory, the clinical samples are subjected to centrifugation to perform RNA extraction. The extracted RNA is used for reverse transcription to form complementary DNA with the help of a thermocycler. For quantification of RNA, reaction mixture containing primers, enzyme mix, and cDNA is added into 96-well reaction plates. The diagnostic results are available within 72–96 h. The diagnosis of SARS-CoV-2 through POC requires M1 Sample Prep Cartridge Kits, which do not require heavy lab equipment. The kit employs filtration-based purification of RNA. The reverse transcription and quantification of RNA are performed by using a battery-operated instrument, the Biomeme Franklin® Real-Time PCR Thermocycler. The results are available within 3–4 h. The multiplex real time PCR reaction takes place in empty Go-strips with three wells, which are sealed with void filling caps before being placed into the thermocycler. The device is connected via Bluetooth to a smartphone to enable clinicians immediate access to the report and analyze data. The diagram was created using BioRender.com.
A limitation of this study is the small number of clinical samples collected at a single site. Multicenter evaluation and outdoor testing of large samples is necessary for a comprehensive analysis of the utility of the Biomeme-thermocycler across a wide range of temperatures and humidities. Although the Biomeme recommended M1 prep kit method of RNA extraction is convenient and rapid, it has the major drawback of recovering only a small amount of nucleic acids, which can lead to false negatives. Despite the availability of several rapid nucleic acid extraction kits, they are not suitable for use with small volumes and complex biological samples. The best way to obtain an accurate POC diagnosis is to optimize RNA extraction kits for the sensitivity and specific performance of Biomeme assays. Furthermore, it is essential to evaluate the sensitivity of the Biomeme thermocycler in the detection of RNA across different specimen types. While the assay detected SARS-CoV-2 RNA from multiple variants, the continuous emergence of variants not tested in this study could alter amplification efficiency.
In conclusion, we developed a single-step RT-qPCR diagnostic assay for the detection of SARS-CoV-2 using the Biomeme Franklin thermocycler. The assay is capable of highly sensitive and specific multiplex detection of the N, E, and S genes of SARS-CoV-2 in clinical samples. Thus, this report aids in the development of a convenient, accurate, and field-deployable diagnostic method for SARS-CoV-2 using a Biomeme thermocycler.