

INTRODUCTION
The coronavirus disease-2019 (COVID-19) pandemic was caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and resulted in millions of infections, poor outcomes in vulnerable populations, and significant global disruption (1, 2, 3). SARS-CoV-2 primarily affects the respiratory system, causing a wide variety of conditions that range from mild cold-like symptoms to severe pneumonia and acute respiratory distress syndrome (ARDS). The latter is a major cause of mortality in patients with severe COVID-19. However, SARS-CoV-2 can affect many other organs as well (4, 5, 6).
SARS-CoV-2 is a novel coronavirus that belongs to the Coronaviridae family. It is a single-stranded, enveloped RNA virus that bears structural similarities to the other lethal coronaviruses, such as SARS-CoV and Middle East respiratory syndrome coronavirus (7, 8). SARS-CoV-2 encodes four major structural proteins, namely, spike (S), envelope, membrane, and nucleocapsid (N). The virus also produces non- structural and accessory proteins that facilitate viral replication and pathogenesis (7, 8).
The S protein mediates SARS-CoV-2 entry into host cells. It has two parts: S1 binds specifically to the host receptor angiotensin-converting enzyme 2 (ACE2), while S2 facilitates membrane fusion between the virus and the host cell (9). ACE2 is expressed by epithelial cells in the lung and nasal mucosae as well as by other tissues (10, 11). The S1/S2 site of the S protein is cleaved by the host enzyme furin, whereas transmembrane protease serine 2 (TMPRSS2) assists in cleaving the S2’ site, facilitating viral entry into host cells (12).
The vital importance of S protein in SARS-CoV-2 infection means that anti-S antibodies can block the infection. Consequently, many anti-COVID-19 vaccines and therapies have been targeted the S protein (13). However, the efficacy of these interventions is complicated by antigenic variation of SARS-CoV-2 at the S protein, particularly its receptor-binding domain (RBD) (14). Indeed, the rapid spread of the virus during the pandemic was greatly facilitated by the emergence of multiple variants, including Alpha, Beta, Gamma, Delta, and Omicron, that bore RBD mutations that increased infectivity and escape from neutralizing antibodies (15, 16). Given the ready mutability of SARS-CoV-2, it is likely that new clinically relevant variants will emerge in the future. To help guide future vaccine design and therapeutic strategies, ongoing research into SARS-CoV-2 induced inflammation is needed.
Dysregulation of immune system is thought to be a key contributor to the most severe COVID-19 cases (17, 18). Specifically, the immune system becomes hyperactivated, which induces tissue damage and can lead to poor outcomes. This hyperactivation can induce a cytokine storm, where pro-inflammatory cytokines such as IL-6, IL-1β, TNF-α, and IFN-γ are produced at excessive levels. The cytokine storm is one of the most prominent features of severe COVID-19 and it overwhelms the homeostatic regulatory mechanisms of the body, particularly those in the lungs (19, 20, 21). It is a major contributor to the development of ARDS.
The immune system can also play beneficial roles in SARS-CoV-2 infection. Specifically, B cells produce protective neutralizing antibodies that help clear the virus and prevent it from infecting host cells (22, 23, 24). These antibodies are key to controlling the infection in most individuals. T cells also play a central role in these protective responses by inducing viral antigen-specific humoral responses and killing virus-infected cells (25, 26, 27). However, severe COVID-19 often associates with T cell exhaustion that is characterized by reduced cytokine production (28, 29). This impairs the ability of T cells to control viral replication and promotes the persistence of infection and inflammation (30, 31, 32).
The global COVID-19 pandemic was eventually curtailed by the development and national administration of effective vaccines against SARS-CoV-2. However, while these vaccines reduce the incidence of severe disease and mortality, breakthrough infections are still occurring, mainly because of the continuous emergence of new SARS-CoV-2 variants, some of which are highly transmissible (e.g. Omicron) (33). Moreover, many individuals continue to suffer from long-lasting post-acute sequelae of SARS-CoV-2 infection (long COVID) that affect multiple organ systems (34) and may be due to ongoing immune dysregulation after viral clearance (32). Thus, it is vital to continue researching the immune mechanisms that underpin COVID-19. The resulting knowledge may help improve vaccine design, inform booster strategies, and lead to more targeted therapies for COVID-19, including for COVID-19 caused by emerging SARS-CoV-2 variants.
Much of the research on antiviral immunity has focused on the adaptive immune system. However, it is increasingly clear that the innate immune system is crucial for host defense against viruses. Of particular importance are the innate lymphoid cells (ILCs), which comprise natural killer (NK) cells and helper ILCs (ILC1, ILC2, and ILC3) (35). ILCs are classified into three major groups based on the transcription factors that drive their development and function, and the cytokines they produce (36, 37). Group 1 ILCs (ILC1s) include NK cells and non-cytotoxic helper ILC1s. They express Eomesodermin (Eomes) or T-bet, produce IFN-γ, and their predominant role is defense from intracellular pathogens, particularly viruses. Group 2 ILCs (ILC2s) express GATA-3, produce IL-5 and IL-13, and associate with type-2 immune responses. Group 3 ILCs (ILC3s) express ROR-γt, produce IL-17 and/or IL-22, and maintain mucosal-barrier integrity.
It was discovered only recently that ILCs not only kill pathogens directly, but they also orchestrate the innate and/or adaptive immune responses to many pathogens, including viruses (35, 38). The latter role is mediated by their secretion of cytokines and other signaling molecules, which shape the responses of other immune cells, including macrophages, dendritic cells (DCs), and T cells (39, 40, 41, 42). This crucial role is facilitated by the fact that unlike adaptive immune cells, ILCs lack antigen-specific receptors and recognize non-specific signals pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) from the tissue environment, particularly at sites of infection or tissue damage (36). Moreover, ILCs are predominantly located at barrier surfaces, which are generally the first points of contact with pathogens, including viruses (43, 44, 45). This strategic location, combined with their functional versatility, positions ILCs to influence the outcome of infection right from the beginning.
These cells recognize viruses or the tissue damage they cause. This causes them to directly destroy viruses: NK cells, for instance, can kill virus-infected cells early in the infection (46). Alternatively, or in addition, the activated helper ILCs, particularly ILC2s, produce immunoregulatory cytokines (36). These functions together with their location mean the ILCs are not only among the first cells to respond to virus infection, they also coordinate other immune cells, including adaptive immune cells, to control the infection. However, ILCs are a two-edged sword: since they play key roles in not only host defense but also tissue homeostasis and repair, ILC deregulation can drive the onset, maintenance, and/or progression of various diseases (36). At present, the exact roles that ILCs play in SARS-CoV-2 infection remain unclear.
This review asks the question, are ILCs mere bystanders in the beneficial and pathological immune responses to SARS-CoV-2, or are they active contributors? We highlight the latest research on the protective and harmful roles of NK cells and helper ILCs in SARS-CoV-2 infection and their potential as therapeutic targets for COVID-19.
SARS-COV-2 ENTRY AND ACTIVATION OF INNATE IMMUNITY
SARS-CoV-2 infection mechanisms
Once the S protein of SARS-CoV-2 binds to ACE2 on target cells, it is cleaved by TMPRSS2, which facilitates membrane fusion and allows the viral-RNA genome to enter the host-cell cytoplasm (47). The viral RNA is translated into viral proteins that are assembled into new virions, which are then released from the infected cells by exocytosis and propagate the infection to neighboring cells.
Since ACE2 is expressed by respiratory epithelial cells and the airways are the most common point of virus-host contact, SARS-CoV-2 infection generally starts in the nasal or lung mucosae. However, because cells in the cardiovascular, gastrointestinal, and nervous systems also express ACE2, the virus can subsequently infect other tissues and organs (48). Such secondary infections contribute to the diverse and sometimes severe clinical manifestations of COVID-19, including myocarditis, acute kidney injury, and neurological disorders (49, 50, 51). This systemic spread of SARS-CoV-2 and its ability to evade natural and vaccine-induced immunity make it a formidable pathogen that is difficult to control.
Activation of innate immunity
After host cells are infected with SARS-CoV-2, they shed viral molecules that bear viral PAMPs (52). The infection also injures the cells, causing them to aberrantly release nuclear or cytosolic proteins that bear DAMPs. These PAMPs/DAMPs are recognized by innate-immune cells via their pattern-recognition receptors (PRRs), which include toll-like receptors, retinoic acid-inducible gene I-like receptors, and nucleotide-binding oligomerization domain-like receptors (53). This recognition of PAMPs/DAMPs by PRR-bearing innate-immune cells activates them and means they are the first immune responders to the infection.
Once activated, the innate immune cells initiate signaling pathways that lead to their production of interferons (IFNs) and pro-inflammatory cytokines. The interferons, particularly type-I (IFN-α, IFN-β) and type-III (IFN-λ), play a central role in activating antiviral responses to SARS-CoV-2. These cytokines signal neighboring cells to enhance their antiviral defenses, including by expressing interferon-stimulated genes (ISGs), which inhibit viral replication and promote viral-RNA degradation (54). However, SARS-CoV-2 suppresses this IFN production and signaling by a variety of mechanisms. This allows the virus to evade the early immune responses and replicate unchecked during the initial stages of infection (55).
The innate immune cells that respond to SARS-CoV-2 infection include macrophages and DCs. They both promote interferon signaling but also contribute to the initial immune response in other ways. Macrophages, particularly those in the lungs, engulf viral particles and release cytokines such as IL-1β and IL-18 that recruit other immune cells to the infection site (56, 57, 58). Moreover, conventional DCs (cDCs) stimulate naïve T cells, thus driving adaptive-immune responses to SARS-CoV-2 (59). However, SARS-CoV-2 can disturb these responses, with pathogenic consequences. Specifically, it can induce alveolar macrophages to overproduce IL-1β and IL-18, thus generating the cytokine storm that characterizes severe COVID-19 (60). Moreover, both cDCs and plasmacytoid DCs, which are major producers of type-I and type-III IFNs in SARS-CoV-2 infection, demonstrate impaired function that may reduce infection control and promote severe COVID-19 (61, 62).
The other major innate immune responders to SARS-CoV-2 are the ILCs. These cells are relatively poorly researched but respond to SARS-CoV-2 as follows.
ROLES OF ILCS IN SARS-COV-2 INFECTION
Role of ILCs in viral infections in general
Multiple studies show that total circulating ILCs are depleted in COVID-19, particularly in severe cases. The research on the protective and pathological roles of ILC1s, ILC2s, and ILC3s in SARS-CoV-2 infections is detailed below (Fig. 1).
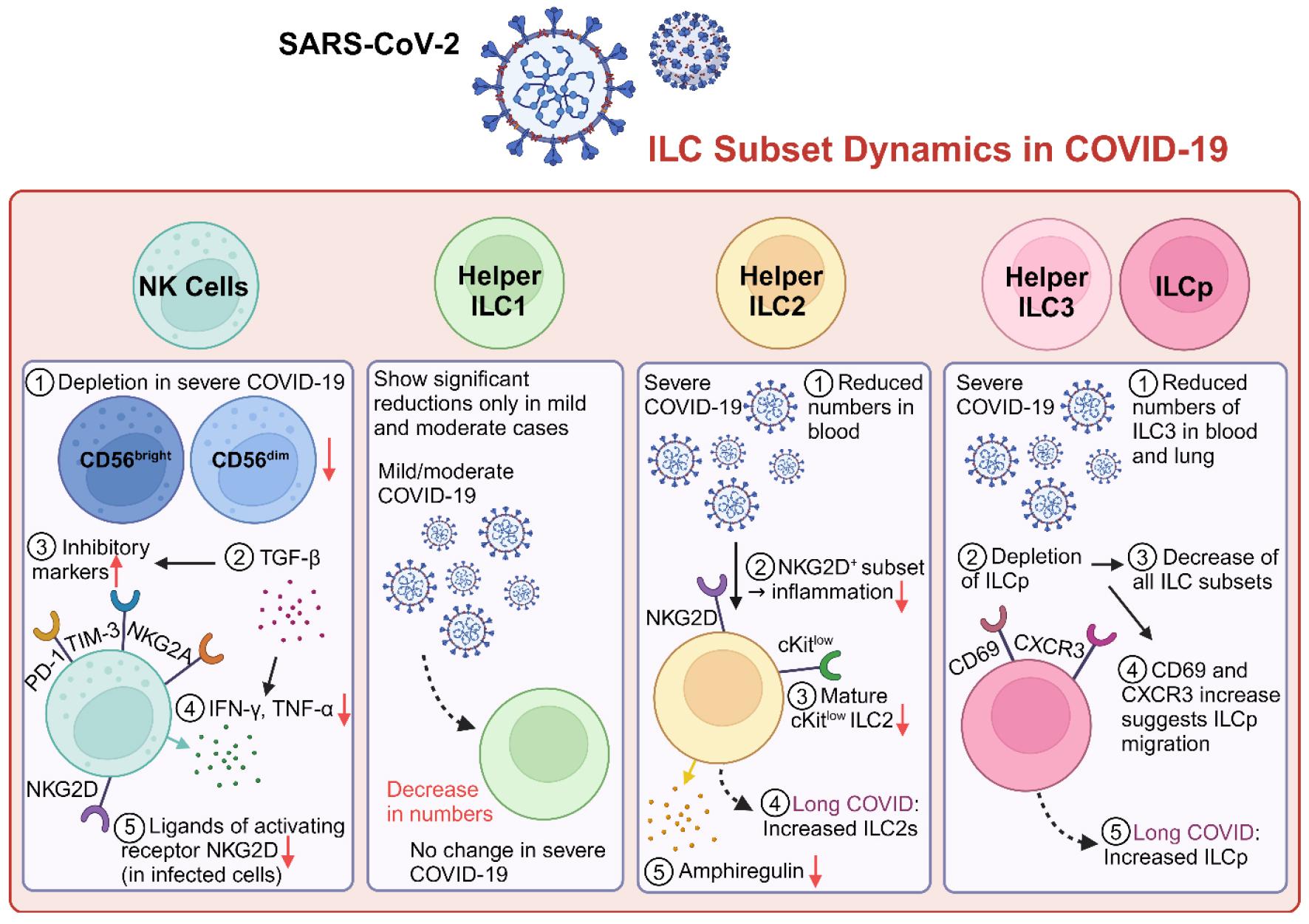
Fig. 1
Dynamics of ILC subsets in COVID-19 patients. In severe COVID-19, NK cells are depleted and exhibit functional exhaustion, marked by increased expression of inhibitory markers (PD-1, TIM-3, and NKG2A). Helper ILC1 numbers decrease in mild/moderate cases but remain stable in severe disease. Severe COVID-19 is associated with reduced ILC2 numbers, diminished Amphiregulin production, and the emergence of an NKG2D⁺ ILC2 subset, which may regulate inflammation. ILC3 depletion affects both blood and lung compartments, potentially impairing mucosal-barrier integrity. ILC progenitors (ILCp) are reduced in severe disease but are elevated in long COVID, where they may contribute to sustained inflammation and tissue repair. These findings underscore the dysregulation of ILC subsets across COVID-19 severity and highlight their potential as therapeutic targets.
Group 1 ILCs
ILC1s include helper ILC1s and NK cells. There is little research on the effect of SARS-CoV-2 infection on the functions and activation of helper ILC1 cells but their numbers are significantly reduced in mild and moderate COVID-19 compared to healthy controls and severe COVID-19 cases (63).
Since NK cells are well-known for their antiviral activities against various viruses, many researchers anticipated that they could also counter SARS-CoV-2 infection. Indeed, COVID-19, which is a proxy for poor infection control, associates with reduced circulating NK cell numbers in multiple observational studies. This is true for both the cytokine-producing (CD56bright) and cytotoxic (CD56dim) NK cell subsets (64, 65, 66, 67, 68, 69). This reduction in NK cells is also observed in the bronchoalveolar lavage fluid (BALF), which reflects the local immune cell landscape in the lung tissue (70, 71). This suggests that the low NK cell numbers in the blood are not due to their migration to the infected and locally inflamed site (71). The possibility that NK cells play antiviral roles in SARS-CoV-2 infection is further supported by the fact that low circulating NK cell numbers, particularly CD107- NK cells and IFN-γ-producing NK cells, correlate with poor clinical outcomes and high levels of inflammatory markers, including C-reactive protein (63, 66, 68, 69, 72, 73). Conversely, convalescent individuals show partial restoration of NK cell numbers (64).
Notably, several studies report that the NK cell population acquires a memory-like phenotype (CD57+ NKG2C+) during COVID-19 convalescence that persists. Studies on other viruses suggest that this phenotype associates with more robust responses on re-infection (65, 66). Thus, SARS-CoV-2 may induce NK cell-mediated immune memory that could be exploited to generate cell-based COVID-19 immunotherapies (74).
Peripheral blood studies show that NK cell responses vary depending on COVID-19 severity. In moderate disease, NK cells are hyperactivated, as shown by higher levels of inflammatory cytokines, cytotoxic markers (e.g. granzyme and perforin), and general activation markers (e.g. Ki-67 and CD69) (75, 76). By contrast, severe disease frequently associates with NK cell exhaustion, as shown by their expression of the exhaustion marker CD39 and lower cytotoxic marker expression (63, 66, 71, 73, 77). This functional exhaustion is also marked by increased expression of immune checkpoint molecules, including PD-1, TIM-3, LAG3, and TIGIT (68, 73, 75, 78). The COVID-19 severity-related changes in NK cell activity reflects the relative balance between the activating and inhibiting receptors on the NK cells (79). For instance, compared to NK cells from paucisymptomatic patients and healthy controls, NK cells in ARDS patients express higher levels of the inhibitory receptor NKG2A but similar levels of the activating receptor NKG2C. Moreover, the high NKG2A expression normalizes after recovery (71, 76, 77). Similarly, COVID-19 associates with downregulation of the activating receptor NKG2D on NK cells (73). In addition, more rapid recovery from COVID-19 associates with greater frequencies of NK cells that express the activating receptor DNAM-1 and are more cytotoxic in ex vivo assays (78). Notably, when NK cells from ARDS patients were treated with an anti-NKG2A monoclonal antibody in vitro, the exhausted cells were reinvigorated (71). Thus, targeting inhibitory receptors on NK cells could have therapeutic potential for severe COVID-19.
Several studies show that SARS-CoV-2 suppresses host-beneficial NK cell activity by various mechanisms. First, SARS-CoV-2 N protein induces NK cells to apoptose in vitro, as shown by their active caspase-3 and CD95 expression, and severe COVID-19 associates with increased expression of these apoptosis markers by circulating NK cells (69). Second, when cells are infected with SARS-CoV-2, its non-structural protein-1 (Nsp1) downregulates ligands for the NK cell-activating receptor NKG2D, which reduces NK cell killing efficiency (80). Third, when epithelial cells are infected with SARS-CoV-2 in vitro, they express and release LLT1, which binds to the receptor CD161 on NK cells. This inhibits NK cell cytotoxicity and cytokine production. Sera from COVID-19 patients were also found to contain increased LLT1 levels (81). Fourth, severe COVID-19 associates with an early (< 2 weeks of infection) increase in circulating TGF-β levels that reduces NK cell expression of genes that relate to cell-cell adhesion and granule exocytosis. Consequently, even though the NK cells bore high levels of cytotoxic effector molecules, their cytotoxicity was strongly suppressed (82). Fifth, severe COVID-19 associates with low serum levels of IL-12, IL-15, and IL-21, which are important for NK cell activity (67).
BALF samples also show that COVID-19 associates with altered lung NK cell activation and increased exhaustion. Thus, in severe COVID-19, lung NK cells express higher levels of the inhibitory receptor NKG2A, the exhaustion marker CD39, and the immune checkpoint molecule PD-1. This recapitulated the findings in circulating NK cells, which confirms that circulating NK cells can act as a surrogate for lung NK cells (71). Moreover, BALF analyses of macaques that were infected with SARS-CoV-2 showed that virus persistence associated with increased alveolar macrophage expression of HLA-E, which interacted with NKG2A on NK cells and reduced their cytotoxicity. By contrast, efficient virus clearance associated with the emergence of memory NKG2Alo NK cells that were resistant to HLA-E-mediated NK cell exhaustion and produced large amounts of IFN-γ that blocked viral propagation in the macrophages (83).
Thus, group 1 ILCs, particularly NK cells, play an important role in SARS-CoV-2 clearance but various mechanisms, including viral evasion strategies and cytokine-mediated suppression, can lead to their exhaustion and dysfunction. This in turn promotes severe COVID-19. Targeting these dysfunction-inducing mechanisms, for example by blocking checkpoint inhibition or NK inhibitory receptors or generating adaptive NK cells, could restore effective anti-viral NK cell responses, thereby improving COVID-19 outcomes.
Group 2 ILCs
ILC2s associate primarily with tissue repair and anti-inflammatory type-2 immune responses. SARS-CoV-2 may activate ILC2s: when mice bearing humanized ACE2 were infected with an S protein-expressing pseudovirus, their mucosal epithelial cells were damaged and released the alarmin IL-25, which in turn activated ILC2s (84). However, multiple studies suggest that while these infection-induced ILC2s are protective in COVID-19, they are downregulated in severe disease. First, observational studies on human blood samples show that severe COVID-19 associates with low ILC2 frequencies (72, 85, 86). Second, ILC2 frequencies correlate negatively with D-dimer levels, which are a marker of COVID-19 clinical severity (87, 88). Third, severe COVID-19 associates with decreased cKitlow ILC2s, which are particularly mature and committed to type-2 cytokine secretion (86, 89). Fourth, ILC2s are elevated in long COVID-19, possibly as a compensatory repair mechanism (90). Thus, by producing type-2 cytokines, ILC2s may modulate SARS-CoV-2 infection and/or disease recovery.
An important ILC2 cytokine in COVID-19 may be amphiregulin (epidermal growth factor-like growth factor), which inhibits inflammatory T cells, terminates inflammatory responses, and induces tissue repair (91). ILC2s in hospitalized COVID-19 patients produce lower levels of amphiregulin than non-hospitalized patients (92). Moreover, Gomez-Cadena et al. (86) showed that severe COVID-19 associates with the emergence of a unique subset of NKG2D+ ILC2s that correlate positively with reduced need for mechanical ventilation. This suggests that ILC2s may dampen inflammation and/or support tissue repair. Notably, they also found that ILC2s from healthy donor blood are readily converted in vitro into NKG2D+ ILC2s by IL-18 alone, and that serum IL-18 levels are significantly higher in severe COVID-19. By contrast, the ILC2-stimulating alarmin IL-33 cannot induce NKG2D expression on ILC2s in vitro, and IL-5 and IL-13, the typical type-2 cytokines of ILC2s, are not elevated in the serum of severe COVID-19 patients. These observations together suggest that NKG2D+ ILC2s may act in a protective manner in severe COVID-19 by producing amphiregulin (86). However, Gomez-Cadena et al. did not examine amphiregulin in their study and further human sample, mouse-infection model, and in vitro ILC2 studies are needed to confirm these findings.
Group 3 ILCs
ILC3s are essential for mucosal barrier integrity. Their roles on COVID-19 are relatively poorly researched because it is difficult to identify blood ILC3 subsets with surface markers only. Nonetheless, one study showed that like the other ILC groups, ILC3s are reduced in the blood of severe COVID-19 patients (72). Thus, SARS-CoV-2 infection may compromise the mucosal barrier support provided by ILC3s, thus promoting severe COVID-19.
Blood ILC progenitors (ILCp)
c-Kit+ CRTH2- ILCs are regarded as ILCps, and they can give rise to all ILC subsets (93). Compared to moderate COVID-19, severe COVID-19 associates with decreased numbers of circulating ILCp. Notably, these cells demonstrate increased expression of the early activation and tissue-resident marker CD69 and decreased expression of the chemokine receptor CXCR3. This suggests that ILCps migrate to the local tissues (85). Interestingly, long COVID associates with increased ILCp frequencies in the total ILC compartment that correlate positively with inflammatory factors, including TNFα, FLT3-ligand, and CXCL1 (94). Thus, ILCps, or the ILCs that differentiate from them, may participate in the sustained immune activation or dysfunction that drives long COVID. These findings together suggest that SARS-CoV-2 infection not only decreases early ILC replenishment, which limits viral clearance, it also induces migration of ILCps to the tissues: these cells then generate the persistent inflammation that results in long COVID-19.
CONCLUDING REMARKS
ILCs appear to promote host defense from SARS-CoV-2 infection since severe COVID-19 associates with low numbers of ILCps and all ILC groups, and convalescence associates with restoration of these numbers, at least in NK cells. The beneficial roles ILCs play are complex and vary per ILC group. NK cells play crucial early antiviral defense roles but exhibit intense activation, functional exhaustion, and poor antiviral efficacy in severe COVID-19, likely because of cytokine-mediated suppression and viral immune evasion. The helper ILCs, particularly ILC2s, orchestrate and dampen immune reactions and protect mucosal tissue but these activities are also diminished in severe COVID-19. Conversely, since ILCps are elevated in long COVID-19, it is also possible that ILCs promote post-acute sequelae.
Therapeutic strategies that correct ILC dysfunction may help improve severe and chronic COVID-19 cases. These strategies could include reactivating NK cells by blocking immune checkpoints or inhibitory NK receptors, modulating cytokines to boost helper-ILC functions, or adoptively transferring memory-like NK cells. To better understand ILC functions in SARS-CoV-2 infection and promote the development of therapies that target their unique contributions in acute and long COVID-19, further research on the ILC changes during infection and recovery is needed.
While the roles of NK cells and ILC2s in COVID-19 have been studied more extensively, the involvement of ILC3s remains poorly understood. Given their critical function in maintaining mucosal integrity and homeostasis, further research is required to elucidate their potential contributions to SARS-CoV-2 infection and disease outcomes.
The question of whether ILCs act as active contributors or passive bystanders in COVID-19 remains open, as most current findings are based on observational and/or correlational studies. Functional investigations are necessary to dissect their precise roles in antiviral immunity, inflammatory regulation, and tissue repair. Such studies will help distinguish whether the observed alterations in ILC subsets during COVID-19 directly influence disease pathogenesis as “active contributors” or represent secondary phenomena as “bystanders.” Bridging this gap between correlation and causation will be critical for determining their therapeutic potential, clarifying their contributions to disease progression and recovery, and informing targeted therapeutic strategies.
