

INTRODUCTION
Autophagy is a conserved lysosome-mediated catabolic process in eukaryotic cells that plays a significant role in antimicrobial defense during infection, serving as a key defense mechanism. The activation of the autophagy/xenophagic process is triggered by complex biological events in response to invading microbes or various inflammatory stimuli. Autophagy plays a crucial role in host defense by coordinating the actions of immune cells, intracellular signaling pathways, and secreted mediators to optimize immune homeostasis during infection (1). Novel therapeutic strategies that modulate autophagy may serve as alternatives for treating infections by enhancing the host's immune response, especially in light of the significant global issue of antimicrobial drug resistance. This review investigates the different types of autophagy that regulate pathogens and the key molecules that coordinate the specific pathways involved.
TYPES OF CANONICAL AUTOPHAGY
Autophagy in mammalian cells can be divided into three types, distinguished by the interaction between the cargo and the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (1). Autophagy can also be classified as nonselective or selective, depending on the nutritional state of the environment. Nonselective autophagy occurs during nutrient deprivation or starvation, whereas selective autophagy is triggered by the recognition of specific cargo, such as an organelle or an invading microbe. These different types of autophagy are described in greater detail below.
Macroautophagy
Macroautophagy, commonly known simply as autophagy, is a catabolic ‘self-eating’ pathway that serves as a critical integration point in numerous biological processes. It also plays a crucial role in maintaining cellular homeostasis, particularly under conditions of stress and infection. In addition, autophagy is a bulk degradation process that removes long-lived proteins and damaged organelles from cells either nonselectively or selectively. It begins with the formation of a double-membraned structure, the autophagosome, that encloses portions of the cytoplasm. Merging of the autophagosome with the lysosome leads to the degradation of its contents (2). The processes that mediate autophagy are orchestrated by more than 30 autophagy-related (ATG) proteins, organized into different functional units (3). Previous studies have provided insights into the origin of the autophagosomal membrane, novel regulators of autophagy, and the mechanisms by which specific intracellular membranes are targeted as autophagy substrates.
Microautophagy
Microautophagy consists of direct phagocytosis by lysosomes and occurs without the formation of an autophagosome, as the autophagic cargo is captured directly at the lysosome membrane via the latter’s protrusion or invagination. Recent studies have proposed two distinct molecular mechanisms for cargo uptake in microautophagy: fission and fusion (4). Fusion-type microautophagy resembles macroautophagy in that it requires the core autophagy machinery and the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Fission-type microautophagy is mediated by endosomal sorting complexes required for transport (ESCRT), which transport the cargo into lytic organelles. Several other forms of microautophagy have been identified as well, including micromitophagy, microreticulophagy, micronucleophagy, macrolipophagy, and endosomal microautophagy. However, microautophagy has been more difficult to study than macroautophagy or CMA, due to limited detection tools and research models (5). A better understanding of how lysosomes target and transport cargo during microautophagy awaits further research.
Chaperone-mediated autophagy
In CMA, a cytosolic chaperone recognizes specific pentapeptide sequences on cytosolic soluble protein substrates, resulting in the direct transport of those proteins to lysosomes for degradation. Among the specific targets of CMA are transcription factors, glycolytic enzymes, calcium, and lipid binding proteins, and vesicular trafficking proteins (6). Cytosolic chaperone proteins include HSC70, which together with its cochaperones recognizes the KFERQ pentapeptide of substrate proteins. The resulting complex binds to the CMA substrate receptor, lysosomal-associated membrane protein (LAMP) 2A, at the lysosomal membrane (7). Following LAMP2A multimerization, HSP90-mediated complex translocation, and stabilization of the complex on the lumenal side of the lysosomal membrane, the substrate protein is translocated into the lumen, where it is eventually degraded (7). CMA is activated in response to oxidative stress, undesirable posttranslational modifications, and significant nutrient depletion. While the regulation of CMA translocation is well understood, the intracellular signaling mechanisms require further study.
AUTOPHAGY PROCESS
Autophagy is a cellular mechanism that facilitates the degradation and recycling of damaged or unnecessary cellular components. The process is divided into several key stages, including initiation, nucleation, elongation, maturation, fusion, and degradation. This section briefly discusses the key molecules and their interactions throughout the macroautophagy process. Fig. 1 depicts an overview of the macroautophagy process, highlighting key components that regulate distinct autophagic pathways.
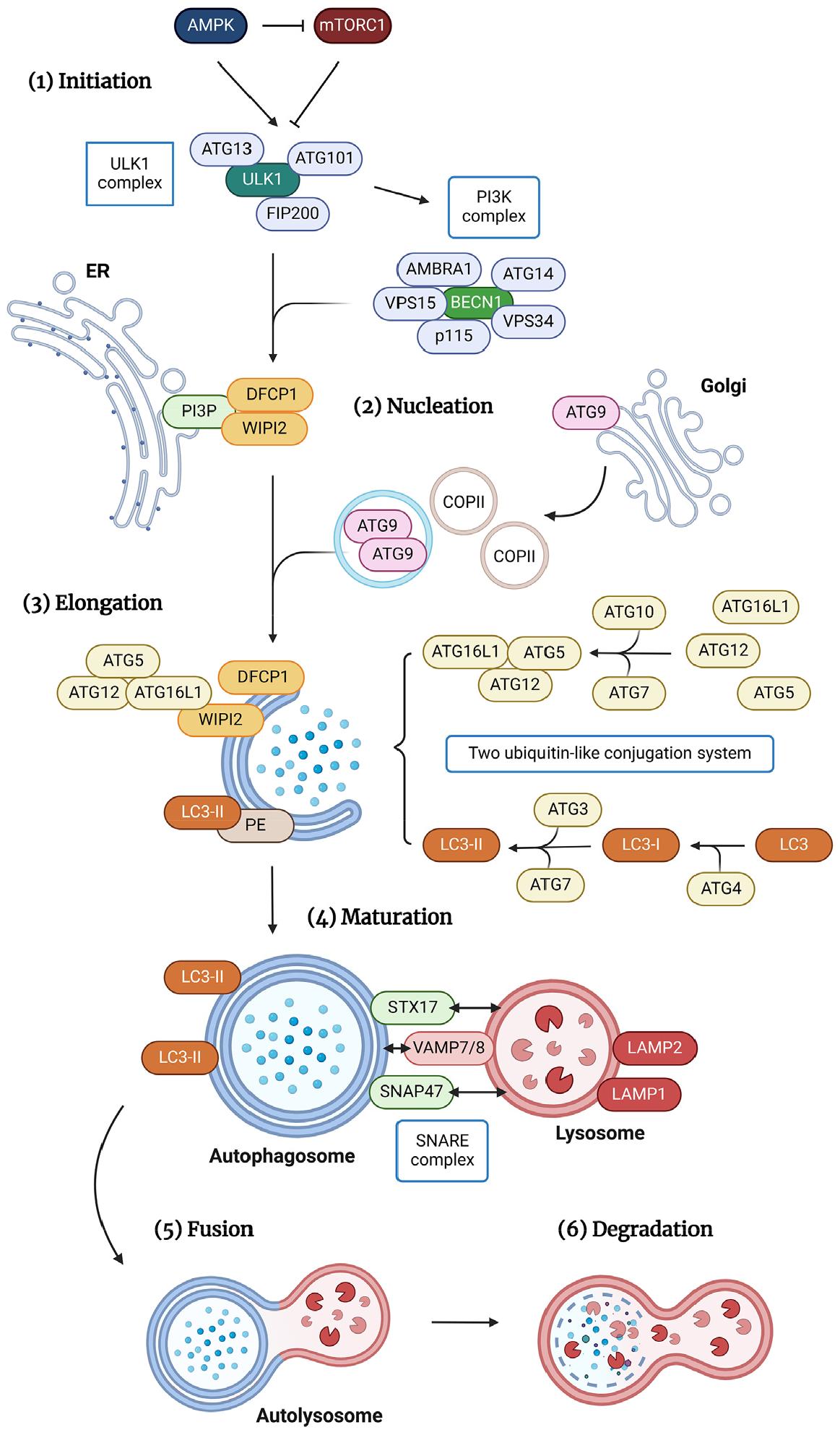
Fig. 1
An overview of macroautophagy pathways depicting key components. (1) Initiation: The ULK1 complex, consisting of ULK1, ATG13, FIP200, and ATG101, initiates autophagy. AMPK and mTORC1 phosphorylation activate and inhibit the ULK1 complex, respectively. Activated ULK1 phosphorylates the PI3K complex, which includes BECN1, AMBRA1, VPS34, VPS15, p115, and ATG14L. (2) Nucleation: Phagophore nucleation begins with the activation of the PI3K complex on the ER membrane, which increases PI3P concentration. Nucleation and elongation occur in PI3P-rich ER regions known as omegasomes, which recruit DFCP1 and WIPI2. ULK1 also recruits ATG9-containing vesicles, COPII vesicles, and unknown membrane components to initiate autophagosome nucleation and elongation. (3, 4) Elongation & Maturation: Two ubiquitin-like conjugation systems coordinate autophagosome elongation and maturation; ATG12 is covalently connected to ATG5/ATG16L1, and LC3 is lipidated with PE. ATG7 and ATG10 covalently conjugate ATG12 with ATG5 and ATG16L1, while WIPI2 binds directly to ATG16L1 and recruits the complex to the membrane. LC3 conjugation to membrane-resident PE generates membrane-bound, lipidated complexes via ATG3. (5) Fusion: The autophagosome and lysosome fuse to form an autolysosome, which degrades cargo. This fusion process requires the SNARE complex, which includes STX17, SNAP47, and VAMP7/8. (6) Degradation: Finally, acidity and lysosomal hydrolases from lysosomes degrade the autophagosome's inner membrane and contents. Their metabolized macromolecules are actively transported into the cytoplasm for recycling or destruction.
Initiation and nucleation
Macroautophagy is initiated by the assembly of the unc-51-like activating autophagy kinase 1 (ULK1)/ATG1 complex, containing ULK1 and the ATG proteins ATG13, FAK family kinase-interacting protein of 200 kDa (FIP200), and ATG101. Activation of the ULK1 complex is regulated by the phosphorylation of two master regulators of cell metabolism: 5′-AMP-activated protein kinase (AMPK) and mechanistic target of rapamycin complex 1 (mTORC1). The ULK1 complex is activated by AMPK phosphorylation and inhibited by mTORC1 phosphorylation. Activated ULK1 is located in the endoplasmic reticulum (ER), where, following its interaction with ATG9 (8), it initiates phagophore nucleation by phosphorylating the phosphoinositide 3-kinase catalytic subunit type III (PI3K) complex, which includes beclin 1 (BECN1), autophagy and BECN1 regulator 1 (AMBRA1), type III phosphatidylinositol 3-kinase (PIK3C3)/vacuolar protein sorting 34 (VPS34), VPS15, p115, and ATG14L (9, 10). Activation of the PI3K complex causes phosphoinositide-3-phosphate (PI3P) to concentrate at the surface of the ER membrane. Specialized ER regions called omegasomes act as PI3P-rich platforms (11) that attract zinc-finger FYVE domain-containing protein 1 (DFCP1) and phosphoinositide-interacting protein 2 (WIPI2) (12) to the phagophore for nucleation and elongation. Recent studies have suggested that the recruitment of ATG9-containing vesicles and coat protein complex II (COPII) vesicles, along with other unknown membrane components, to the ULK1 complex provokes autophagosome nucleation and elongation (13, 14). These structures thus collectively serve as platforms for autophagosome biogenesis. Their further study will provide insights into the early stages of membrane formation.
Elongation and maturation
Two ubiquitin-like conjugation systems, consisting of ATG12 covalently linked with ATG5/ATG16L1 and microtubule- associated protein light chain 3 (LC3)/ATG8 lipidated with phosphatidylethanolamine (PE), are required for the coordinated action that leads to the elongation and transformation of the single-layer membrane into the double-membrane vesicle known as the autophagosome. DFCP1 and WIPI2 recruit these two systems to the elongation site (12). Initially, ATG12 is covalently conjugated with ATG5 and ATG16L1 via ATG7 and ATG10; then WIPI2 binds directly to ATG16L1, recruiting the ATG12–ATG5–ATG16L1 complex to the membrane (12, 15). ATG3-mediated conjugation of LC3 to membrane-resident PE leads to the formation of membrane-bound, lipidated structures. In this process, the covalent attachment of a PE molecule to LC3 begins with the cleavage of the terminal arginine residue of LC3 by ATG4 protease, revealing a glycine residue at the C terminus that allows the formation of LC3-I. Subsequently, the proteolyzed protein is recognized and processed by two enzymes: the E1-activating enzyme ATG7 and its own E2-like enzyme, ATG3. Ultimately, the E3-like ATG12–ATG5–ATG16L1 complex facilitates the definitive ligation of LC3 to PE, thereby converting LC3-I into LC3-II, the signature molecule of autophagosome membranes (10, 16).
Fusion and degradation
Finally, the autophagosome merges with the lysosome, giving rise to an autolysosome where the engulfed cargo undergoes degradation. This critical fusion event requires the participation of the SNARE complex (17), which includes autophagosome-localized Q-SNARE STX17. The latter binds to VAMP8, a lysosome-localized R-SNARE, via an autophagosome- localized Q-SNARE, SNAP29 (18). However, a recent study reported that the STX17-SNAP47–VAMP7/VAMP8 SNARE complex, rather than SNAP29-containing SNAREs, is the default SNARE complex governing autophagosome-lysosome fusion, responsible for both selective and nonselective autophagy (19).
In addition to SNAREs, a group of proteins engaged in fusion also function as constituents of the cellular machinery associated with other transport mechanisms that converge on the lysosome/vacuole (10). Among these proteins are the homotypic fusion and vacuolar protein sorting (HOPS)/class C VPS tethering complex (20), small GTPase RAB7 (21), and several members of the ESCRT protein family (22, 23). Both the VPS34–BECN1–UVRAG complex (24, 25) and LAMP1 (26) are required for lysosome–autophagosome fusion. The specific functions of these proteins within the complex network of cellular processes, including autophagy-related fusion events and other crucial pathways that direct cellular components toward the lysosome/vacuole, remain to be unraveled.
After fusion with the lysosome/vacuole, the inner membrane of the autophagosome and its contents are degraded by lysosomal hydrolases and the acidic environment. Then the catabolized macromolecules are actively transported from these organelles into the cytoplasm, where they can be recycled or further degraded.
AUTOPHAGY IN INFECTION
Autophagy across infection stages
Autophagy is considered a critical defense mechanism against various infections and plays a dynamic role throughout the bacterial infection process. During the early stages of infection, autophagy helps identify and sequester pathogens that enter the host through a mechanism known as xenophagy (27). Autophagosomes then engulf the pathogen, fusing with lysosomes to degrade it, allowing for early infection control. Autophagy activation at the initial stage appears crucial for inhibiting the progression of the acute phase of infectious diseases (28, 29, 30).
As the infection progresses, autophagy may act as a defense mechanism and a regulator of immune responses, combating pathogens while avoiding collateral damage to host tissue (31). During viral infection, autophagy triggers an innate immune response by collaborating with pattern recognition receptor signaling to induce interferon production (32). Once the pathogen has been cleared or contained, autophagy continues to support the host's immune response by removing cellular debris and damaged organelles caused by the infection (33). It facilitates the resolution of inflammation and tissue repair, thereby ensuring efficient recovery from infection. Autophagy is also recognized for its role in regulating inflammation through the inhibition of the inflammasome, a multiprotein complex associated with the pathogenesis of various inflammatory and autoimmune diseases (34, 35). However, some pathogens exploit the autophagy machinery to survive and establish a replication niche in host cells. For example, Legionella pneumophila and hepatitis C virus (HCV) were shown to induce autophagosome formation but inhibit phagolysosomal fusion, resulting in microbial replication (32, 36). Mycobacterium tuberculosis activates the mTOR pathway to actively inhibit autophagy (37), while Listeria monocytogenes manipulates LC3-associated phagocytosis (LAP) to form spacious Listeria-containing phagosomes (SLAPs) (38). From this perspective, host-directed therapies that enhance autophagy or target pathogen mechanisms that bypass autophagic pathways have the potential to be novel therapeutic strategies in infectious diseases.
In the later stages of infection, autophagy may help host defense by facilitating antigen processing and presentation, thereby empowering adaptive immune responses (32). For example, human immunodeficiency virus (HIV) type 1 is targeted to autophagosomes, where it is processed by autophagic machinery before being presented on MHC class II molecules (39). Autophagy is also involved in the transport and processing of extracellular antigens for cross-presentation by MHC class I molecules (40). However, chronic pulmonary inflammation can lead to persistent unregulated autophagy, which may worsen lung injury (41) This suggests that a regulated autophagy response is crucial for maintaining immune system homeostasis, especially in the context of inflammation management.
Autophagy and pathogen control
Autophagy, specifically xenophagy, functions as an effective defense mechanism against intracellular pathogens. Many intracellular pathogens, including Streptococcus pyogenes, M. tuberculosis, L. monocytogenes, and Salmonella Typhimurium, have been identified as effective autophagy targets (42). However, these pathogens also have evolved strategies to adapt successfully against autophagy mechanisms within host cells (43, 44).
Although autophagy primarily targets intracellular pathogens, it also contributes to the defense against extracellular pathogens by activating neutrophil function (45) and facilitating antigen presentation (46). Autophagy promotes neutrophil degranulation and the release of neutrophil extracellular traps, which can trap and kill extracellular pathogens (45). Autophagosomes form in MHC class II-positive dendritic cells and fuse with late endosomal compartments to transport cytoplasmic proteins for MHC class II presentation (32).
In summary, autophagy serves as a crucial defense mechanism against both intracellular and extracellular pathogens. However, its effectiveness can be undermined by sophisticated pathogen evasion and exploitation strategies, emphasizing the intricate interplay between host defense and microbial survival mechanisms. While the role of autophagy against bacterial infections has been extensively studied, more research is needed to understand the precise mechanisms of autophagy pathways involved in infections across different stages and pathogen types.
SELECTIVE AUTOPHAGY
Selective autophagy differs from nonselective autophagy in that it targets the degradation of specific cellular components, such as damaged organelles (mitophagy), protein aggregates (aggrephagy), and pathogens (xenophagy). This is accomplished by specific cargo receptors known as selective autophagy receptors (SARs), which contribute to autophagosome formation by interacting with LC3/ATG8 family proteins via the sequence motifs making up the LC3-interacting region (LIR) (47). An in-depth review of selective autophagy is provided in previous studies (48, 49). In the following sections, we briefly discuss the mechanisms and key features of several types of selective autophagy associated with infectious conditions. Fig. 2 illustrates the specific SARs that participate in the selective autophagy pathways.
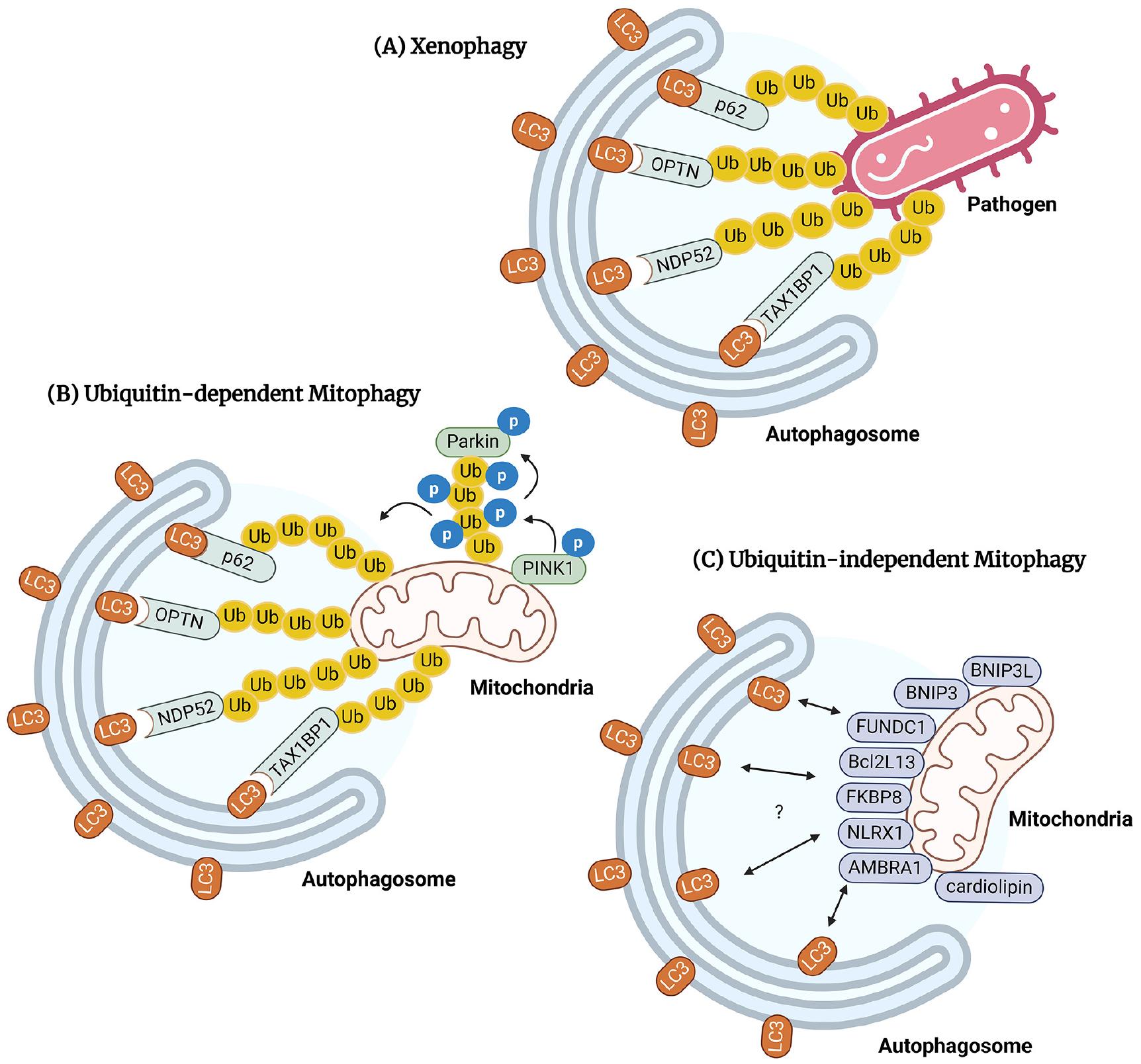
Fig. 2
SARs involved in xenophagy and mitophagy. (A) Xenophagy engulfs pathogens in the cytosol or vacuole. SARs such as p62, OPTN, NDP52, and TAX1BP1 mediate xenophagy by selectively recognizing and labeling pathogens with ubiquitin chains. SARs collaborate with LC3 to activate autophagy and eliminate pathogens through lysosomal clearance. (B) Ubiquitin- dependent mitophagy recognizes and activates autophagic degradation of damaged or excess mitochondria. PINK1 accumulates in damaged mitochondria and recruits Parkin ligase to activate mitophagy. Mitophagy recognizes damaged mitochondria using cargo receptors such as p62, OPTN, NDP52, and TAX1BP1, similar to xenophagy. (C) Ubiquitin-independent mitophagy involves mitochondrial outer membrane SARs, including NIX/BNIP3L, BNIP3, FUNDC1, Bcl2L13, FKBP8, NLRX1, AMBRA1, and cardiolipin.
Xenophagy
Xenophagy targets cytosolic or vacuolar pathogens by enveloping them in autophagosomes and breaking them down within lysosomes. It is therefore a critical part of the innate immune defense system. Xenophagy is mediated by various SARs, such as p62/SQSTM1 (50), OPTN (51), NDP52 (52), and TAX1BP1 (53), which play crucial roles in selectively recognizing and labeling pathogens with ubiquitin chains. During this process, galectins bind to damaged membranes (54) and have been implicated in the inhibition of pathogens such as M. tuberculosis, group A Streptococcus, and hepatitis B virus (HBV) (55, 56, 57). However, a recent study found that galectin-3, -8, and -9 play only limited roles in resistance to acute M. tuberculosis, S. Typhimurium, or L. monocytogenes infections (58). During infection, multiple SARs interact with LC3/GABARAP to facilitate the interaction between the recognized pathogen and the autophagy machinery, ultimately leading to lysosomal clearance and effective pathogen elimination.
Studies involving the deletion of various autophagy genes both in vitro and in vivo have provided valuable insights into the role of xenophagy in controlling bacterial replication. For instance, mice with myeloid-derived cell-specific ATG5 deletion (Atg5fl/fl LysM-Cre) succumb to M. tuberculosis infection due to the hyperactivated pathological inflammation induced by neutrophil infiltration (59). Moreover, a recent study demonstrated that robust depletion of ATG16L1 or ATG7 leads to increased M. tuberculosis growth, enhanced necrosis, and heightened host susceptibility in mice (60). Future studies exploring the in vivo function of ATG genes should thus consider the nature of those infections. In addition, numerous pathogens have evolved strategies to evade xenophagy or manipulate the xenophagic machinery for their own benefit. For example, group A Streptococcus expresses the cysteine protease SpeB, which can degrade cargo receptors such as p62 and NRB1 (61). L. pneumophila, an intracellular pathogen, produces an effector protein, RavZ, to deconjugate LC3/ATG8 from ATG3 and ATG7, thus preventing xenophagy (62). Viruses such as HBV, HIV, and influenza A virus also possess unique xenophagy evasion tactics (63, 64, 65). Nonetheless, much remains to be learned about the molecular mechanisms by which individual pathogens manipulate the host xenophagy process to establish and maintain their infections in host cells.
Mitophagy
Mitophagy selectively targets and stimulates the autophagic degradation of damaged or excessive mitochondria. It has garnered recent attention following the discovery of a link between mitophagy and the pathogenesis of Parkinson’s Disease (66). In mammals, Ser/Thr kinase PTEN-induced putative kinase protein 1 (PINK1) and the E3 ubiquitin ligase Parkin are key proteins that regulate mitochondrial quality (67). PINK1 rapidly accumulates in damaged mitochondria, causing the recruitment of the Parkin ligase (68, 69, 70), which then mediates the ubiquitination of voltage-dependent anion channel 1 (VDAC1) and the recruitment of p62 to activate mitophagy (71). Prohibitin 2, an inner mitochondrial membrane protein, is a vital receptor involved in mitophagy via a PINK-Parkin-dependent pathway (72). Like xenophagy, mitophagy requires SARs, such as NDP52 and OPTN, to recognize damaged mitochondria (73).
In addition to ubiquitin-dependent mitophagy, a ubiquitin-independent pathway has been identified that involves several SARs located on the mitochondrial outer membrane, including NIX/BNIP3L (74), BNIP3 (75), FUNDC1 (76), Bcl2L13 (77), FKBP8 (78), NLRX1 (79), AMBRA1 (80), and cardiolipin (81). However, their specific molecular mechanisms are so far unknown. The interactions of the different processes in the mitophagy pathway are also poorly understood.
Recent studies suggest that mitophagy closely regulates the innate immune response to infection (82). In a polymicrobial sepsis model, PINK1-Parkin pathway dysfunction was shown to increase host susceptibility and inflammasome activation (83). Impaired mitophagy leads to the accumulation of damaged mitochondria and thus potentially to mitochondrial- DNA-mediated inflammation (84, 85). Pathogens such as L. monocytogenes have evolved to manipulate mitophagy to improve their survival, by targeting the mitophagy receptor NLR family member X1 (NLRX1) (79). Other pathogens induce mitophagy to suppress innate immune responses and maintain infection. For example, influenza A virus proteins such as PB1-F2 and nucleoprotein stimulate mitophagy to inhibit innate immunity (86, 87), while the lytic replication of human herpes virus 8 is promoted by NIX-mediated mitophagy induction (88). These observations demonstrate the complexity of mitophagy-mediated innate immunity control in terms of the type of pathogen, host cell responses, and disease models. Understanding the intricate role of mitophagy under infectious conditions will aid the development of mitophagy-targeted therapies for both known and emerging pathogens. Moreover, because mitophagy and xenophagy share several cargo receptors, such as p62, OPTN, NDP52, and TAX1BP1, insights into their crosstalk, as well as their relative contributions to different immune stages, may improve our understanding of the key roles of selective autophagy-mediated host defenses against pathogens.
NONCANONICAL AUTOPHAGY
Canonical autophagy proceeds in a well-defined sequence of steps, including formation of the autophagosome for lysosomal degradation. The pathways of noncanonical autophagy differ from the canonical pathway in terms of the molecular machinery involved and the cargo type targeted for degradation. The following section briefly discusses one such pathway, LAP, including the mechanisms and key features that contribute to inflammation and infection.
LC3-associated phagocytosis (LAP)
In LAP, autophagy components are directly conjugated with LC3 to phagosomal membranes via the recruitment of Rubicon to activate the PI3K complex, a process that replaces the formation of the autophagosome’s characteristic double-membrane structure (89). Specifically, upon infection, membrane-bound receptors, such as Toll-like receptors (TLRs), dendritic cell-associated C-type lectin-1 (dectin-1), and Fc-gamma receptors (FcγRs), on phagocytes activate the LAP pathway (90, 91), which induces the recruitment of the NADPH oxidase 2 complex and is stabilized by Rubicon, an indispensable component of the BECN1-VPS34 autophagy complex and LAP maturation (92). NADPH oxidase-derived reactive oxygen species (ROS) lead to LC3 lipidation, resulting in the formation of the LAPosome, which fuses with lysosomes to eliminate pathogens through phagolysosome complexes (90).
Recent studies have shed light on the molecular mechanisms governing LAP pathway activation in the context of infections. During LAP, the integration of lipidated LC3 into endolysosomal membranes is facilitated by the WD repeat-containing C-terminal domain of ATG16L1 (93). ATG16L1 is recruited by V-ATPase, via K490, and participates in the conjugation of LC3/ATG8 to single endolysosomal membranes (94). Histoplasma capsulatum infection triggers LAP activation by involving the Dectin-1-Syk pathway and NLRX1. The interaction between NLRX1 and Tu translation elongation factor (TUFM) causes their association with autophagic proteins ATG5–ATG12, leading to the formation of LAPosomes. The NLRX1–TUFM complex-dependent pathway also plays a vital role in LAP-mediated MAPKs-AP-1 activation, which is essential for the cytokine response to H. capsulatum (95). During infection with L. monocytogenes, the integrin ITGAM- ITGB2, also known as Mac-1 or CR3, acts as the receptor responsible for initiating the activation of antimicrobial immune responses by LAP (96). Engulfment and cell motility protein 1 (ELMO1) also actively participates in LAP by facilitating LC3 accumulation and enhancing antibacterial responses during enteric infection (97). Studies in animal models have demonstrated the vital role of LAP activation in enhancing antimicrobial defenses in various infection scenarios. A study of a zebrafish model of invasive Aspergillosis showed that autophagy-related responses, including LAP, augment host defenses (98). These and similar compelling findings underscore the potential of LAP pathways as promising host-directed therapies for combating infections by drug-resistant pathogens.
AUTOPHAGY AND IMMUNOMETABOLISM
During an infection, macrophages and other immune cells undergo metabolic reprogramming, a stepwise adaptation that allows host cells to meet the bioenergetic requirements of biosynthetic pathways (99, 100). However, some pathogens exploit host immunometabolic responses by utilizing the metabolites released by host cells for their own survival and proliferation (100). A significant shift towards glycolysis during acute infections links to inflammatory responses and the development of trained immunity (101), which in turn provides innate memory, enhancing nonspecific protection against secondary infections via the epigenetic reprogramming of myeloid cells. The metabolic shift towards increased glycolysis in trained monocytes depends on the Akt-mTOR-hypoxia-inducible factor 1-alpha (HIF-1α) pathway (101). Although mTOR activation inhibits autophagy, exogenous lactate treatment of human macrophages ultimately attenuates the glycolytic shift and maintains oxidative phosphorylation in these cells. Lactate also promotes autophagy and enhances the intracellular clearance of M. tuberculosis (102). These data suggest that early immunometabolic responses to microbial stimuli are characterized by increases in glycolytic flux and lactate production, mediated by mTOR-HIF-1α signaling. Concurrently, these early responses are associated with enhanced inflammatory responses, the activation of autophagy, and heightened bactericidal function during infection.
In the later phase of infection, intracellular metabolism redirects enhanced mitochondrial oxidative phosphorylation (OXPHOS) to the generation of mitochondrial ROS (mtROS), which act as antimicrobial effectors and play a crucial role in activating bactericidal functions (103). Recent studies have identified a dual role of mtROS in antimicrobial responses during infection (104): regulating autophagy pathways and maintaining homeostasis under stress conditions such as infection and cellular damage (104). However, excessive mtROS can damage host cells and cause pathological inflammation. mtROS levels are regulated by TNF signaling, which promotes cellular glutamine uptake and succinate accumulation, leading to mtROS generation and necrosis (105). The complex I inhibitor metformin prevents TNF-mediated mtROS production, thereby enhancing bacterial clearance (105).
In addition, during the early phase of TLR4 stimulation with LPS, there is an increase in aerobic glycolysis and a decrease in OXPHOS. Conversely, TLR2 activation in monocytes by Pam3Cys-Ser-(Lys)4 (Pam3CSK4) increases oxygen consumption, OXPHOS, and lipid metabolism. Pharmacological inhibition of mitochondrial respiratory chain complex I reduces cytokine production and phagocytosis in Pam3CSK4-stimulated monocytes (106). M. tuberculosis requires fatty acids and cholesterol for its survival, due to its lipid-rich cell wall (107). Blocking host fatty acid β-oxidation (FAO) activates antimicrobial killing mechanisms through mtROS bursts, which promote macrophage NADPH oxidase activity and xenophagy (108). However, one study showed that FAO metabolism in murine alveolar macrophages is necessary to inhibit foam cell formation (109). Autophagy has an intricate relationship with the immunometabolic status of infected cells, although the precise mechanisms have not been fully elucidated.
Emerging evidence highlights that the interplay between autophagy and immunometabolism is essential for shaping immune defenses at various stages of infections, affecting both pathogen clearance and immune cell function. Dysregulation of autophagy by pathogens can disrupt the balance between host immune and metabolic pathways, potentially impairing immune responses and increasing disease susceptibility. Further research into these interactions within the context of specific pathogenic infections will provide valuable insights into how targeting metabolic and autophagic pathways can enhance immune responses and improve disease management. This understanding could drive the development of innovative therapeutic strategies regulating autophagy and metabolism to optimize infection outcomes.
AUTOPHAGY AND DRUG-RESISTANT INFECTION
The emergence of drug-resistant pathogens has forced the urgent development of new therapeutics to control infectious diseases. Previous studies have shown that intravenous immunoglobulin-induced autophagy was critical in bactericidal effects against drug-resistant Escherichia coli and Pseudomonas aeruginosa in human neutrophils (110). Furthermore, autophagy has been shown to alleviate organ damage and improve the survival rate of methicillin-resistant Staphylococcus aureus (MRSA)-induced septic mice by inhibiting Th1 and Th17 responses (111). Recently, several autophagy-activating molecules were found to have antibacterial properties against multidrug-resistant mycobacterial infections. V46, a novel resveratrol derivative, demonstrated effective antimicrobial activity against multidrug-resistant Mycobacteroides abscessus through autophagy and transcription factor EB activation (112). Dimethyl itaconate-mediated autophagy activation has also been shown to enhance host defense against multidrug-resistant M. tuberculosis infection. (113). Further research is necessary to elucidate the specific mechanisms connecting autophagy activation to the clearance of multidrug-resistant mycobacteria. However, the regulation of autophagy presents a promising therapeutic target for the eradication of multidrug-resistant bacteria.
CONCLUSION
This study examines the detailed mechanism of autophagy potentially activated during infection. Current research focuses on the development of innovative host-directed therapies to fight the emergence of multidrug-resistant bacteria. The regulation of macroautophagy and selective autophagy, including xenophagy, mitophagy, and noncanonical autophagy such as LAP, may play crucial roles in preventing the emergence of new infectious diseases and managing existing intractable infectious diseases through the development of novel autophagy-activating agents. Further research is necessary to elucidate the intricate mechanisms and roles of essential molecules that govern autophagy pathways in the defense against pathogens.
