

INTRODUCTION
폴리오바이러스는 3개의 혈청형(Type 1, 2, 3)을 가지며, 혈청형간 교차면역이 없어 백신제조 시 3개의 혈청형 항원을 각각 제조하여야 한다. 폴리오백신은 1955년 Salk에 의해 주사용 불활화 사백신(Inactivated poliomyelitis vaccine, IPV)이 개발되었고, 1962년 Sabin에 의해 첫 경구용 생백신(Oral poliomyelitis vaccine, OPV)이 허가를 획득하였으며 (1), 두 형태의 백신이 전 세계적으로 가장 많이 사용되고 있다. 그러나 야생 폴리오에 의한 감염이 발생하지 않는 지역에서 경구용 생백신을 계속해서 접종하면 오히려 백신의 약독화된 바이러스에 의해 폴리오(Vaccine-derived poliovirus, VDPV)가 유행하여 백신 관련 마비성 소아마비(Vaccine-associated paralytic poliomyelitis, VAPP)가 발생할 수 있다 (2). 따라서 다시 변이가 일어나지 않는 불활화 백신을 사용해야만 폴리오의 완전한 박멸을 이룰 수 있다. 세계보건기구(World Health Organization, WHO)는 감염원이 줄고 있는 상황에서 가장 위험한 요인이 될 수 있는 야생형 솔크주(Salk strain)보다 약독화된 사빈주(Sabin strain)를 사용하여 불활화 폴리오 바이러스 백신을 제조하고 철저하게 관리할 것을 권장하고 있다 (3, 4).
생물학적 제제는 백신, 혈장유래 제품, 항독소 등과 같이 유기체 또는 유기체 유래물질을 포함하는 것으로, 이러한 물질은 배양, 정제 및 불활성화 같은 제조 과정에서 변형될 가능성이 있다. 생물학적 제제는 제조에 사용되는 재료와 방법의 다양한 특성으로 인해 각 로트의 활성과 안전성을 지속적으로 유지하는 것이 상당히 어렵다. 그리고 생물학적 활성과 안정성은 단순한 물리화학적 방법으로 측정할 수 없으므로 생물학적 제제의 역가시험은 다른 생물학적 표준품과 비교하여 평가되어야 한다. 따라서 생물학적 제제의 품질관리를 위해서는 표준품의 표준화 및 품질관리가 중요하다 (5).
표준품은 영국 국립 바이오 의약품 표준화 연구소(National Institute for Biological Standards and Control, NIBSC)와 유럽 의약품 품질관리청(EU/EDQM(European Directorate for the Quality of Medicines & HealthCare))에서 확립 및 관리하는 국제표준품과 각 국가 규제기관에서 확립하여 관리하는 국가표준품이 있다 (6, 7, 8). NIBSC에서는 사빈주 불활화 폴리오백신(Sabin strain inactivated poliomyelitis vaccine, sIPV)의 국제표준품(17/160)을 2018년 확립하였다 (9). 기존의 솔크주 폴리오백신 국제 표준품(3차 국제표준품 NIBSC Code. 12/104)은 단위를 D-antigen Unit (DU)로 사용하였으나 (10), 사빈주 불활화 폴리오백신의 국제표준품은 항원성의 차이로 Sabin D-antigen Unit (SDU)이라는 새로운 단위가 도입되었다 (11). 2020년 국내에서도 첫 사빈주 불활화 폴리오백신이 허가되었으며 그 개발 때부터 품질관리를 위한 국가표준품의 필요성이 제기되었다. 이에 2019년 국가표준품 후보물질을 제조하였고 2020년 본연구로 이에 대한 D-항원함량 값을 설정하여 국내 첫번째 사빈주 불활화 폴리오백신 국가표준품을 확립하고자 하였다.
MATERIALS AND METHODS
사빈주 불활화 폴리오백신 국가표준품 후보물질의 제조
WHO에서 공급하는 국제표준품은 실험실표준물질의 보정을 위한 목적으로 연간 소량만 공급하고 있어 안정된 품질관리를 위해 국가표준품의 확립이 필요하다 (6, 12). 또한 제조사에서 사용하는 in-house 표준품의 경우, 솔크주 국제표준품으로만 보정한 것으로 사빈주 국제표준품으로 보정한 SDU 값이 필요하였다. 이에 2019년 용역기술사업(19202생물제259, 2019년 사빈주 불활화 폴리오백신 1차 국가표준품 후보물질 제조)을 진행하여 국가표준품 후보물질을 ㈜LG화학에서 총 6980 바이알 제조하였다.
사빈주 불활화 폴리오백신 국가표준품 후보물질(이하, 국가표준품 후보물질) 제조를 위해서는 3가지 타입의 불활화 사빈주 폴리오 바이러스 항원물질이 필요하다. 항원물질 생산에 사용된 세포주 및 바이러스주는 ㈜LG화학에서 특성분석을 실시하여 항원물질의 생산에 적합함이 확인되었으며, 사용된 바이러스주, 세포주에 대한 정보는 Table 1과 같다. 아프리카 녹색 원숭이 신장세포인 Vero 세포주를 계대배양한 후 사빈주 폴리오 바이러스를 각 타입별로 접종하여 바이러스를 대량 생산하였다. 회수한 바이러스를 농축 및 정제하고 포르말린을 이용하여 불활화하여 제균여과한 후 각 타입별 항원을 제조하였다.
Table 1.
The information of Cells and Virus strains
| Cell line | WHO Vero cell seed (ECACC, Lot No. 87-10) | |
|---|---|---|
| Virus strain | Sabin strain type 1 | LSc, 2ab-KP2 |
| Sabin strain type 2 | P712, Ch, 2ab-KP2 | |
| Sabin strain type 3 | Leon 12,a1b-KP3 | |
각 타입별 항원은 국가표준품 후보물질 제조를 위해 사용되었다. 사빈주 불활화 폴리오백신의 D-항원함량 분석 시 표준품의 희석 및 손실량 등을 고려하여 1형 폴리오 항원은 50 DU/mL, 2형 폴리오 항원은 80 DU/mL, 3형 폴리오항원은 160 DU/mL 이 되도록 희석하고 제균여과하여 국가표준품 후보물질을 제조하였다. 제조된 국가표준품 후보물질을 유리바이알에 0.65 mL씩 충전하고 고무마개 및 알루미늄 캡으로 밀봉하여 –60℃ 이하에 보관하였다.
사빈주 불활화 폴리오백신 국가표준품 후보물질의 품질평가
제조 후 액상의 국가표준품 후보물질과 동결하여 –60℃ 이하에 보관하였던 국가표준품 후보물질을 실온에서 녹여 품질평가를 실시하였다. 시험방법은 대한약전의 일반시험법 및 제조소에서 개발한 특정시험법에 따라 수행하였다. 액상의 동결 전 후보물질로 단백질 함량시험, 잔류 소혈청 알부민 함량시험, 포름알데히드 함량시험, 엔도톡신 시험, 무균시험을 수행하였으며, -60℃ 이하에 동결 후 실온에 녹인 후 성상시험, pH 측정시험, 확인시험, D-항원함량시험을 수행하였다. 확인시험은 D-항원함량시험과 함께 진행하였으며, D-항원함량 시험의 경우 3회 시험으로 3반복하여 총 9회의 시험결과를 평균하여 결과처리 하였다.
사빈주 불활화 폴리오백신 국가표준품 후보물질의 공동연구
참여기관 설정
국가표준품 후보물질의 D-항원함량 설정을 위하여 다기관 공동연구를 계획하였다 다기관 공동연구의 경우 기관별 실험실 환경의 차이와 시험장비의 차이로 시험 결과에서 오차가 많이 나타나므로, 숙련도 오차의 최소화를 위해 이전에 폴리오백신의 D-항원함량 시험을 수행한 경험이 있는 제조소, 시험검사기관 및 국제연구소인 ㈜LG화학, 메타바이오㈜, 국제백신연구소를 선정하였으며 식품의약품안전평가원 내 표준품 관리부서인 바이오의약품연구과도 함께 공동연구를 진행하였다. 또한 백신의 시험검정을 하는 백신검정과와 같은 업무를 하고 있는 일본국립감염병연구소(The National Institute of Infectious Diseases, NIID)도 함께 참여하였다.
프로토콜 마련
국제적으로 공인된 폴리오백신의 D-항원함량시험은 형-특이 항원의 함량을 ELISA (Enzyme-linked immunosorbent assay) 방법으로 측정하는 것이다 (13, 14). ELISA 시험은 항체 특이성 및 시약 반응성에 따라 영향을 받으며, 각기 다른 단클론항체 또는 다클론항체 기반의 ELISA 절차를 사용하는 경우 D항원 함량의 직접적인 비교는 불가능 하다. 그리하여 자체 ELISA 방법을 개발하여 형-특이 D항원 함량을 측정하도록 하며, 이 때 사용되는 자체 표준품은 제품 개발 초기 단계에 확정하고 국제표준품으로 교정하게 되어있다 (6). 폴리오백신의 국제표준품은 WHO/NIBSC에서 분양하는 솔크주 “International Standard for IPV 12/104”, 사빈주 “WHO International Standard Sabin Inactivated Polio Vaccine (sIPV) 17/160”과 EU/EDQM에서 분양하는 “Poliomyelitis vaccine (Inactivated) BRP batch 3”이 있으나, 우리의 국가표준품 후보물질은 솔크주와 사빈주에 대한 국제표준품을 모두 갖춘 NIBSC 국제표준품(NIBSC code:12/104, 17/160) (Supplement 1)을 사용하여 D-항원함량을 구하기로 하였다.
공동연구용 프로토콜은 국가표준품 후보물질의 제조소 D-항원함량 시험법을 참고하여 공동연구에 맞게 작성하였다.
① 샘플의 설정
NIBSC 국제표준품 솔크주(12/104), 사빈주(17/160), 국가표준품 후보물질, 시험적합성을 확인하기 위한 ㈜LG화학의 in-house 표준품과 정도관리물질 총 5개를 공동연구용 샘플로 설정하였다.
② 샘플의 시작 희석배수 설정
ELISA를 이용하는 시험법으로 표준품 대비 검체의 농도를 구하고자 할 때 시험하고자 하는 표준품과 검체의 농도를 비슷하게 전희석하고 이를 단계희석 후 결과를 도출하게 된다. 그러나 이번 공동연구에는 5종류의 샘플, 3개의 타입, 그리고 8구간으로 희석하면서, 만들어야 하는 희석검체 수를 줄이기 위해 타입별 전희석 배수를 모두 40배로 설정하였다.
③ 항체 및 시약
프로토콜에 사용된 항체 및 시약은 허가받은 국내 사빈주 불활화 폴리오백신의 국가품질검사 시 시험에 사용한 것과 동일한 것으로 사용하였다. 그 이유는 사빈주 불활화 폴리오백신의 국가품질검사 시 기존 제조사의 표준품을 사용했을 때와 새로 확립한 국가표준품으로 D-항원함량시험 할 때, 항체 및 시약 차이에 의한 결과 차이를 줄이기 위해서였다.
시험에 사용한 항체는 코팅항체(Polyclonal), 검출항체(Monoclonal)는 ㈜LG화학에서 만든 것을 사용하였으며, 2차항체는 Peroxidase AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (H+L), Jackson ImmunoResearch Cat. No. 114 036 146을 사용하였다.
다기관 공동연구 수행
선정된 6개 실험실에 마련된 공동연구용 프로토콜과 수행에 필요한 4회분의 국가표준품 후보물질 및 시약 등을 정해진 보관온도를 유지하여 공급하였다. 우선 각 기관에서 예비시험(1회)을 수행한 다음 그 결과를 받아 검토 후 공동연구진행에 문제가 없는 지 확인한 후 본시험 3회를 타입별로 수행하였다. 96 well plate에 타입별 코팅항체를 well 당 100 ㎕씩 분주하여 25℃, 14시간 이상 흡착시킨 후, 3회 반복하여 세척하고 Blocking buffer를 well 당 100 ㎕씩 분주하여 37℃ 1시간 동안 반응 후, well을 3회 세척한다. 국가표준품 후보물질과 함께 2종 국제표준품, in-house 표준품, 정도관리 물질은 튜브에서 40배 희석하고 이후 8회 2배씩 단계희석하여 well 당 100 ㎕씩 분주하여 37℃, 1시간 반응하고 well을 세척 3회 한다. 검출 단클론항체를 well 당 100 ㎕씩 분주하여 37℃, 2시간 동안 반응시키고, 2차 항체를 well 당 100 ㎕씩 분주하여 37℃, 1시간 반응한 후, TMB(3,3’,5,5’-tetramethylbenzidine) 용액을 well 당 100 ㎕씩 주입한 후 차광하여 실온에서 10분간 발색 후 1 N 황산을 100 ㎕씩 분주하여 반응 정지시킨다. 이후, 10분이내에 650 nm를 보조파장으로 하여 450 nm에서 흡광도 측정하고 측정한 OD값을 제공한 파일에 기입하여 제출한다. 시험 모식도는 Fig. 1과 같다.
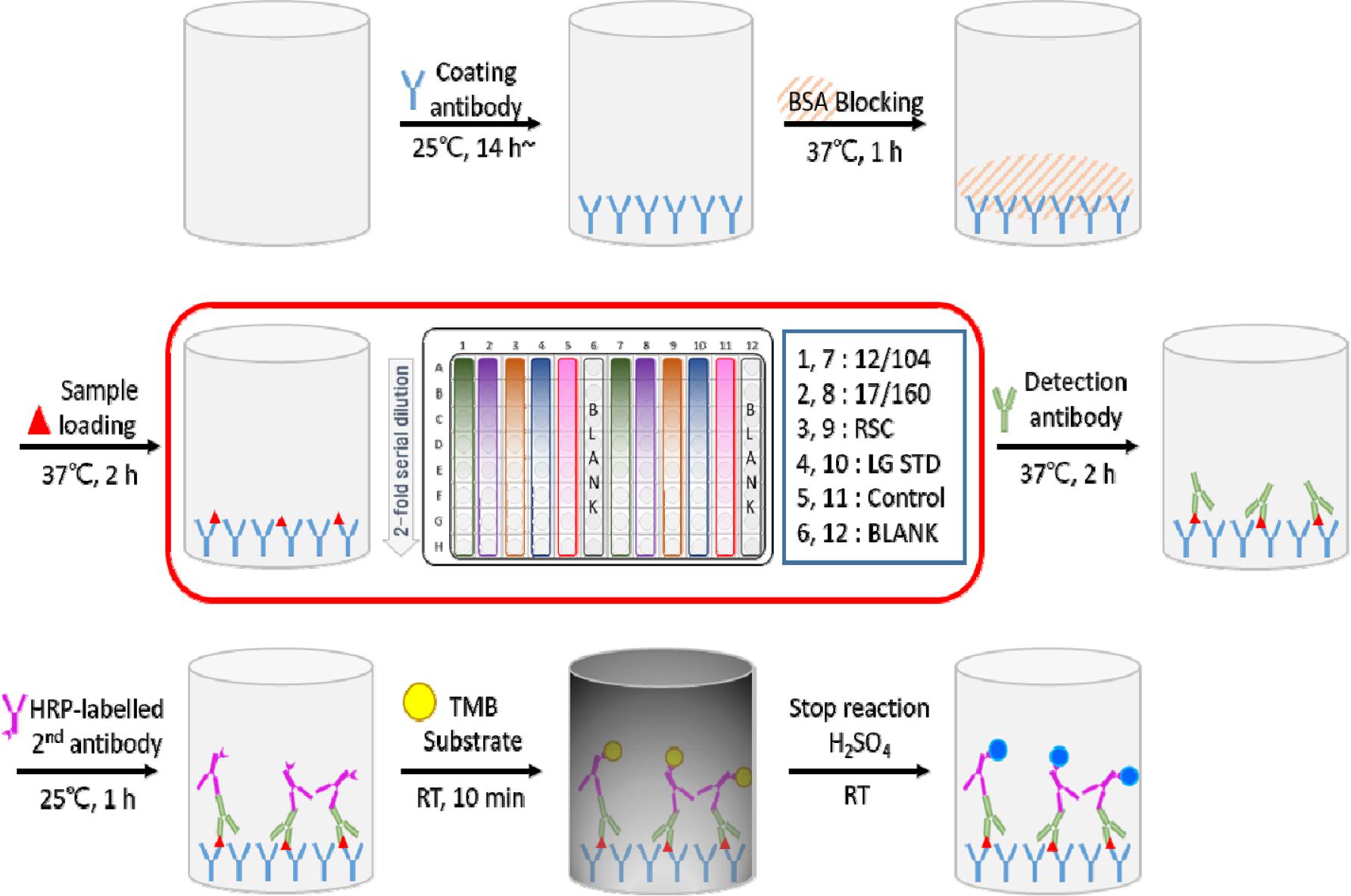
공동연구 결과 데이터 및 통계분석
평행선 시험 수행
6개 참여 기관의 ELISA Raw data을 OD 값으로 받아 CombiStats 프로그램을 사용하여 분석하였다. 유럽약전 Chapter 5.3에 제시된 바와 같이 평행선 모델(Parallel line models: 표준품의 용량-반응 직선과 만나기 위하여 검체의 용량-반응 직선이 수평으로 이동하는 거리로 검체의 상대역가를 계산)을 사용하여, Assess statistical validity by analysis of variance (ANOVA) 통계방법을 이용하여 Regression(회귀)는 significant (p<0.05), non-parallelism(비평행성) 과 non-linearity(비직전성)는 not significant (p>0.05)일 때 도출되는 함량으로 결과를 분석하였다. 이때, 국제표준품 12/104 대비 후보물질의 DU/mL 값을, 17/160 대비 후보물질의 SDU/mL 값을 각각 분석하였다. 시험적합성 확인을 위해 제조사의 in-house 표준품 대비 정도관리 물질의 값이 허용기준에 적합한지를 분석하였다. 허용기준은 3명의 시험자가 총 14회 시험한 결과를 이용하여 예상함량값을 설정한 후 ± 3SD 로 허용범위를 설정하였다(Table 2).
Table 2.
Acceptance criteria of controlled substance
| Controlled substance | ||
|---|---|---|
| Expected content (DU/mL) | Acceptance Criteria (± 3SD) | |
| Type1 | 9.3 | 6.2~ 12.4 |
| Type2 | 16.9 | 10.9 ~ 22.8 |
| Type3 | 35.6 | 24.7 ~ 46.4 |
Table 2-1.
The result of test suitability for each of laboratory
다기관 공동연구 결과 통계분석
시험적합성에 만족하고 시험 프로토콜대로 시험을 수행한 기관에서 도출된 D-항원함량의 예비값들을 모수적방법(Z-점수)을 이용하여 통합역가의 분포가 정규분포를 구성한다는 가정하에 이후 기하평균을 이용하여 통합역가를 산정하였다. 기하평균의 95% 신뢰구간은 GMk ± t(α/2; n-1) x GSEk, t(0.975:n-1)=자유도 (n-1)인 t분포 97.5% 백분위 계산식을 이용하여 구하였다.
국가표준품 후보물질의 안정성 시험연구
식품의약품안전처의 의약품 등의 표준품안정성지침에 따라 D-항원함량값으로 장기 안정성시험을 수행하였다. 후보물질의 보관온도 조건인 –60℃ 이하에서 제조소에서 자사표준품으로 수행하던 0, 3, 6개월에 이어 식약처에서 국제표준품으로 11, 12, 18개월의 장기안정성 시험을 통해 후보물질의 안정성을 확인하였다. 또한 제조 후 3개 온도 조건((5 ± 3)℃, (25 ± 2)℃, (37 ± 1)℃)에서 0, 2, 4주에 가속 안정성시험을 수행하였다. –60℃ 이하의 보관 조건을 갖는 국가표준품 후보물질에 대한 관리 또는 사용 지침을 제시하기 위하여 Freezing/Thawing에 따른 D-항원함량을 검토하고자 하였다. 제조 후 –60℃ 이하에 동결 보관하였던 국가표준품 후보물질을 D-항원함량 시험을 위해 해동한 Freezing/Thawing 1회 시료와 실온에서 2시간 동안 해동한 후 다시 –60℃ 이하에서 밤샘 동결을 추가 2회 반복한 Freezing/Thawing 3회 시료에 대해서 D-항원함량 시험을 실시하였다.
RESULTS
국가표준품 후보물질의 품질평가
제조 후 동결하기 전 후보물질의 품질을 평가하기 시험한 결과, 단백질 함량은 20 ㎍/mL 미만, 잔류 소혈청 알부민 함량과 포름알데히드 함량은 각각 0 ㎍/mL, 0 mg/mL 값을 보여 공정 중 정제가 잘 되었음을 보였다. 또한 엔도톡신과 무균시험결과도 모두 적합으로 외부 미생물이 감염되지 않음을 확인하였다. 영하 60℃ 이하로 동결 후 다시 해동하여 성상과 pH, 확인, 함량시험을 수행하였으며, 이때, 확인시험은 D-항원함량시험으로 특정 항체에 반응함을 확인하였다. D-항원함량시험 결과 제조 시, 예상함량과 유사하게 나타났음을 확인하였다. 품질시험 결과는 Table 3과 같다.
Table 3.
The results of quality test of Sabin Inactivated Polio Reference candidates
| Sample Condition | Test Method | Acceptance Criteria | Result | |
|---|---|---|---|---|
| Before Freezing | Protein content | ≤ 100 μg/mL | ≤ 20 μg/mL | |
| Bovine serum albumin | ≤ 1 μg/mL | 0 μg/mL | ||
| Formaldehyde content | ≤ 1 mg/mL | 0 mg/mL | ||
| Endotoxin test | ≤ 10 EU/mL | < 0 μg/mL | ||
| Sterility test | No growth | Pass | ||
| After Freezing | Appearance | Colorless, transparent solution | Pass | |
| pH | Information onlyb | 6.7 | ||
| Identity testa | Type 1 | Be confirmed | Pass | |
| Type 2 | Be confirmed | Pass | ||
| Type 3 | Be confirmed | Pass | ||
| D-Ag contentc | Type 1 | ≥ 30 DU/mL | 46 DU/mL | |
| Type 2 | ≥ 48 DU/mL | 84 DU/mL | ||
| Type 3 | ≥ 96 DU/mL | 195 DU/mL | ||
표준품 후보물질의 D-항원함량
폴리오백신의 품질관리 시험 중 하나인 D-항원함량시험 수행에 필수적인 표준품을 사용하기 위해서는 표준품의 타입별 표시 함량이 통계적으로 타당하게 설정되어야 한다. 이에 제시된 시험방법으로 제조사 등 6개의 기관에서 각각 3회씩 수행하였다. 기관별로 온 결과를 Combistat으로 분석하여 D-항원함량으로 계산하였고 이 중 정도관리 물질의 값을 확인한 결과, 기관 C의 경우, 총 9회 결과 중 7회가 허용기준에 포함되지 않아 해당 기관의 결과는 최종 함량 도출에 사용하지 않았다. 또한 기관 F의 경우 측정장비의 흡광도 파장과 항체 보관온도를 프로토콜에서 제시한 것과 다르게 시험하여 이 또한 최종 함량 도출에 사용하지 않았다(Table 2-1). 즉 D-항원함량의 설정에 4개기관 3회반복 결과 총 12개의 결과를 분석하였다. 각 타입별 기하평균은(GMT)은 DU/mL 값의 경우 Type 1 46.96, Type 2 129.29, Type 3 262.38으로 나타났으며 표준편차(SD)는 Type 1 1.20, Type 2 1.23, Type 3 1.35 로 나타났다. SDU/mL 값의 경우 Type 1 47.59, Type 2 106.03, Type 3 192.80으로 나타났으며 표준편차(SD)는 Type 1 1.08, Type 2 1.07, Type 3 1.08 로 나타났다. 또한 타입별 DU/mL의 경우, 95% 신뢰구간은 Type 1 46.84 ~ 47.09, Type 2 129.15 ~ 129.43, Type 3 262.15 ~ 262.60을 나타냈으며, 타입별 SDU/mL의 경우, 95% 신뢰구간은 Type 1 47.59 ~ 47.64, Type 2 105.98 ~ 106.07, Type 3 192.75 ~ 192.85로 나타났다(Supplement 2). 본 연구에서는 정도관리물질을 사용하여 1차적으로 적합하지 않은 결과를 분석에서 제외하였으므로 함량 산정에 쓰인 몇몇 값들이 통계적으로 이상 값을 보인다 할 지라도 모든 값을 포함하여 함량을 산정하였다. 국가표준품 후보물질의 최종 기하평균 D-항원함량 역가는 DU 및 SDU 로 타입 1, 2, 3형 별로 47 DU/mL, 129 DU/mL, 262 DU/mL, 48 SDU/mL, 106 SDU/mL, 193 SDU/mL로 산정하였다(Table 4).
후보물질의 안정성 연구
제조된 국가표준품 후보물질의 실시간 장기안정성 시험을 수행하여 후보물질의 안정성을 확인하였다(Fig. 2). 당초 9개월에 국제표준품 대비 실시간 안정성 시험을 수행하고자 하였으나 국제표준품의 배송지연으로 11개월에 시험을 수행하여, 후보물질의 보관온도 조건인 - 60℃에서 0, 3, 6, 11 12, 18 개월에 후보물질의 안정성을 확인하였고 18개월 동안 D-항원함량에 유의미한 차이가 없는 것을 확인하였다. 0개월차 안정성시험은 release 시험 결과로 갈음하였다. 다만 6개월까지는 제조소에서 자사 표준품 대비로 DU/mL 값을 구하였으나, 이후부터 18개월까지는 식약처에서 국제표준품(12/104) 대비 DU/mL 값을 구하여 안정성시험을 수행하였고, SDU/mL 값은 6개월부터 도출하였으며 제조소와 식약처 모두 국제표준품(17/160)을 기준으로 값을 도출하였다(Table 5).
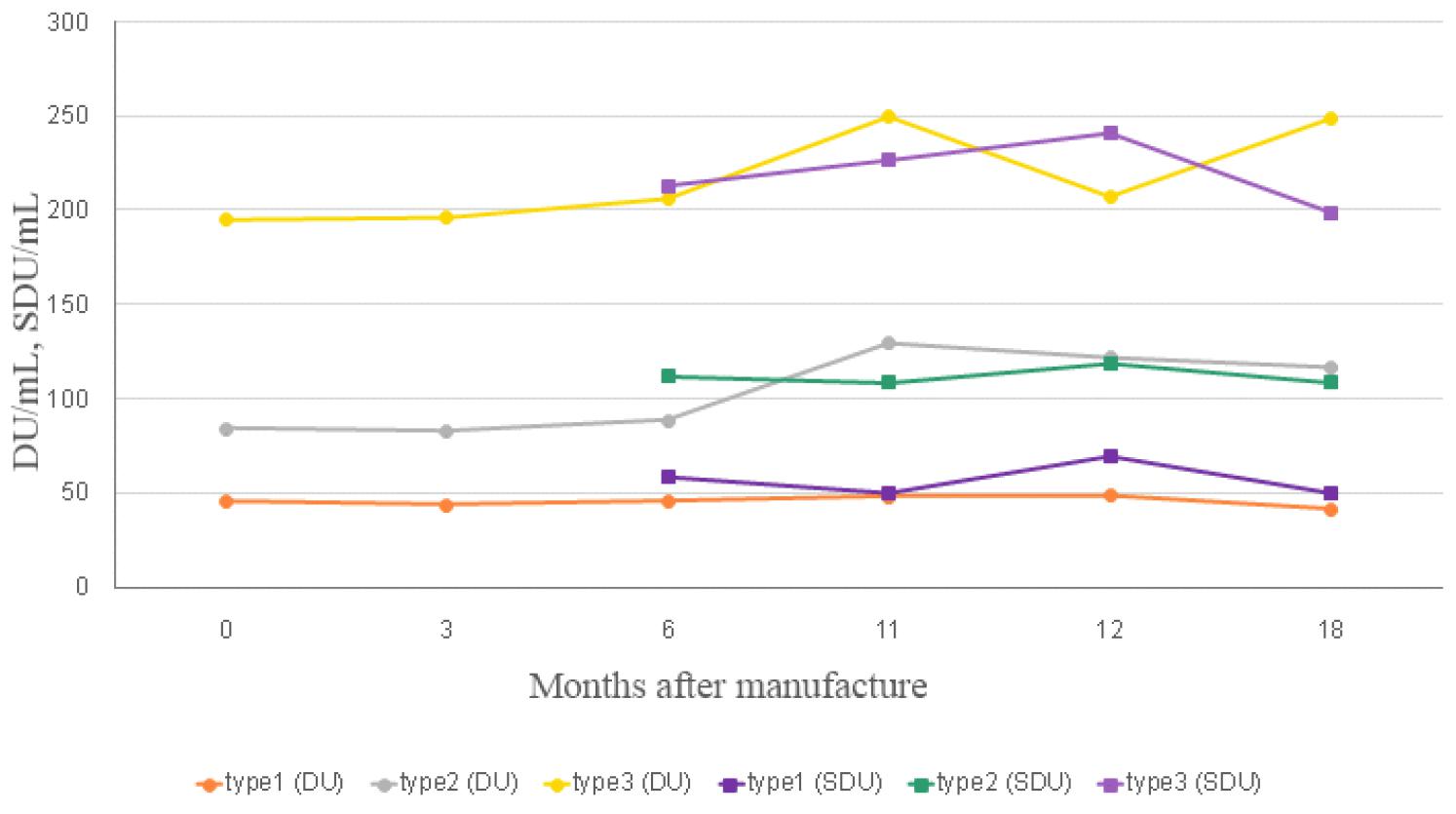
Table 5.
The results of Long-term stability test for reference standard candidates
| Test Method | Long-term Stability (Month) | ||||||
|---|---|---|---|---|---|---|---|
| 0a | 3b | 6 | 11a | 12 | 18 | ||
| Appearance | pass | pass | pass | pass | pass | pass | |
| D-antigen Contents (DU/mL) | Type 1 | 46 | 44 | 46 | 48 | 48 | 41 |
| Type 2 | 84 | 83 | 87 | 129b | 122 | 117 | |
| Type 3 | 195 | 196 | 206 | 250 | 208 | 249 | |
| D-antigen Contents (SDU/mL) | Type 1 | - | - | 59 | 50 | 70 | 50 |
| Type 2 | - | - | 112 | 109 | 119 | 109 | |
| Type 3 | - | - | 213 | 226 | 241 | 199 | |
제품의 사용방법 및 보관 등을 고려하여 Freezing/Thawing test와 가속시험을 수행한 결과, Freezing/Thawing 3회 반복에 따른 D-항원함량 검토를 위하여 Freezing/Thawing 1회 했을 때 D-항원함량 결과를 기준으로 회수율(%)을 계산하였고, 그 결과 Supplement 3과 같이 국가표준품 후보물질에 대하여 Freezing/Thawing 3회 반복에 따른 D-항원함량의 변화가 없음을 확인할 수 있었다. 가속 안정성 시험 결과는 Supplement 4와 같다. 국가표준품 후보물질은 (5 ± 3)℃ 및 (25 ± 2)℃ 조건에서 4주간 성상, pH 및 D-항원함량 시험에서 모두 기준에 만족하는 것을 확인하였다. (37 ± 1)℃ 조건에서는 성상이나 pH 변화는 보이지 않았지만, D-항원함량의 감소가 관찰되어 본 국가표준품 후보물질을 (37 ± 1)℃ 조건에서 보관하는 것은 적합하지 않음을 확인할 수 있었다.
DISCUSSION
WHO에서는 이미 기존에 솔크주 국제표준품으로 사빈주 백신의 품질관리는 적절치 않다고 발표하였으며 (11), 이에 WHO는 2018년 사빈주 국제표준품을 확립하여 타입 1, 2, 3 별로 새로운 단위인 SDU를 사용하여 100 SDU/mL, 100 SDU/mL, 100 SDU/mL으로 역가를 산정하여 분양하고 있다 (9). 다만, 사빈주 국제표준품 확립 이전에 개발 및 시험법을 확립한 백신의 경우, NRA의 승인을 받아 DU 단위를 사용할 수 있다고 말하고 있다 (14).
이에 본 연구에서는 국제표준품 솔크주 12/104와 사빈주 17/160 을 사용하여 다기관 공동연구를 통하여 국가표준품 후보물질의 D-항원함량 값을 DU 및 SDU 로 타입 1, 2, 3형별로 47 DU/mL 129 DU/mL, 262 DU/mL, 48 SDU/mL, 106 SDU/mL, 193 SDU/mL로 산정하였으며 2021년 국가표준품(MFDS-B-19-001)으로 등록을 마쳤다. 연구결과를 살펴보면 사빈주 국가표준품 후보물질을 솔크주 국제표준품 12/104로 측정한 DU값의 GCV%가 타입별로 20%, 23%, 35%를 나타내, 17/160으로 측정한 SDU값의 GCV% 타입별 8%, 7%, 8% 보다 높게 나타남을 알 수 있었다. 즉 결과의 일관성이 SDU-SDU가 높게 나타남을 알 수 있었다. 제조사측에서 제조 시에 설정한 DU 함량값이 공동연구에서 나온 DU 값과 다르나, 이는 사용한 표준품이 다르기 때문이며, 공동연구를 통해 표준품의 역가를 산정할 때 흔히 나타나는 결과이다.
또한 좀 더 신뢰있는 결과를 도출하기 위하여 공동기관의 수 및 시험의 수를 증가하여 수행하고 싶었으나, 시험에 사용하는 모든 항체 등 시약을 제조사의 제공으로 공급되어야 하여, 그 분량이 충분하지 않았고 사빈주 불활화 폴리오백신이 국내 최초로 허가받은지 얼마되지 않아 경쟁사인 제조기관들이 공동연구에 참여할 수 없어 아쉬웠다. 그럼에도 최대 많은 결과를 D-항원함량 산정에 포함시키려 하였다. 결과분석 시 기관 C의 경우 시험적합성 결과가 다른 기관들과 상이하여 결과분석에 사용할 수 없었으며, 기관 F의 경우 항체의 희석 및 보관 그리고 측정 흡광도 파장(630 nm 사용)을 제공된 공동연구용 프로토콜과 상이하게 진행되어 그 결과가 다른 기관들과 큰 차이가 있음을 확인되어 결과 분석에 사용할 수 없었다.
통계분석은 이상치를 제외하고 설정하는 것이 타당하나, 기존에 시험에 대한 숙련도가 높은 기관을 선정하여 공동연구를 진행하였고 시험적합성 등을 확인하여 적절치 않은 시험기관의 결과를 제외하였으므로, 통계적으로 이상치 값을 나타내 제외해야 하는 결과일지라도 그 이상치 값이 크지 않은 경우 모두 포함하여 통합역가를 산출하였다. 또한 설정된 D-항원함량을 근거로 국가표준품 후보물질을 이용하여 상용 사빈주 불활화 폴리오백신(유폴리오주)의 역가를 측정하였을 때 모두 허가 기준에 적합한 결과를 얻음으로써 국가출하승인 시 국가표준품으로 사용 가능성을 확인하였다. 또한 현재는 상용 사빈주 백신에 SDU 단위가 없으나, 향후 제조사가 보다 안정적인 SDU값을 설정해야 할 때, 국가표준품을 사용하여 국제적이고, 표준화된 품질관리를 수행할 수 있을 것이다.
SUPPLEMENT
Supplement 1.
International reference standard of poliomyelitis vaccine
| 3rd International Standard for IPV (cIPV) | Sabin Inactivated Polio Vaccine (sIPV) | |
|---|---|---|
| NIBSC Code | 12/104 | 17/160 |
| Unit | DU/mL | SDU/mL |
| Type1 | 277 | 100 |
| Type2 | 65 | 100 |
| Type3 | 248 | 100 |
Supplement 2.
The D-Ag result from each laboratory used for statistical analysis
| DU/mL | Type1 | Type2 | Type3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Lab./Assay | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| A | 43.02 | 45.34 | 48.15 | 141.31 | 116.31 | 111.46 | 312.26 | 224.02 | 205.13 |
| B | 42.26 | 69.44 | 40.61 | 101.31 | 180.69 | 127.4 | 262.51 | 417.49 | 296.25 |
| D | 41.59 | 44.62 | 41.52 | 121.39 | 115.54 | 122.96 | 239.76 | 247.87 | 303.12 |
| E | 48.28 | 66.78 | 41.41 | 149.95 | 191.07 | 103.34 | 292.61 | 344.06 | 125.97 |
| GMa | 46.96 | 129.29 | 262.38 | ||||||
| SDb | 1.20 | 1.23 | 1.35 | ||||||
| Lower 95% | 46.84 | 129.15 | 262.15 | ||||||
| Upper 95% | 47.09 | 129.43 | 262.60 | ||||||
| SDU/mL | Type1 | Type2 | Type3 | ||||||
| Lab./Assay | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| A | 45.69 | 46.48 | 50.53 | 97.11 | 106 | 96.71 | 189.77 | 192.51 | 201.18 |
| B | 54.91 | 51.92 | 50.3 | 111.39 | 116.89 | 108.45 | 197.68 | 205.86 | 206.02 |
| D | 43.69 | 45.27 | 42.76 | 105.93 | 101.33 | 95.09 | 187.88 | 183.12 | 196.44 |
| E | 47.49 | 49.54 | 44.11 | 110.11 | 118.76 | 107.57 | 207.6 | 195.66 | 155.97 |
| GMa | 47.59 | 106.03 | 192.80 | ||||||
| SDb | 1.08 | 1.07 | 1.08 | ||||||
| Lower 95% | 47.54 | 105.98 | 192.75 | ||||||
| Upper 95% | 47.64 | 106.07 | 192.85 | ||||||
Supplement 3.
The results of Freezing and Thawing for candidate
| Freezing/Thawing number | Type 1 | Type 2 | Type 3 | |||
|---|---|---|---|---|---|---|
| 1 | 3 | 1 | 3 | 1 | 3 | |
|
D-Ag content (DU/mL) | 47.0 | 48.0 | 84.3 | 84.4 | 198.5 | 210.4 |
| 45.9 | 46.2 | 83.4 | 86.3 | 199.1 | 201.9 | |
| 43.5 | 44.5 | 86.3 | 87.2 | 203.9 | 198.0 | |
| Average (DU/mL) | 45.5 | 46.2 | 84.6 | 86.0 | 200.5 | 203.4 |
| % RSDa | 4.0 | 3.8 | 1.8 | 1.6 | 1.5 | 3.1 |
| Recovery (%)b | 100 | 102 | 100 | 102 | 100 | 101 |
