

INTRODUCTION
Chikungunya virus (CHIKV), a positive-sense, single-stranded RNA virus belonging to the genus Alphavirus and family Togaviridae, poses a major public health concern, with millions of clinical cases reported worldwide (1). Its transmission, primarily through Aedes mosquitoes, contributes to its widespread dissemination in human populations. The virus was first sequenced in 1953, and four genotypes were identified: Indian Ocean (IO), West African (WAf), East/Central/South African (ECSA), and Asian.
Molecular diagnostics have proven valuable in diagnosing CHIKV infections in patients. Real-time PCR is commonly used during the acute phase of infection to detect viral RNA in plasma or serum within 2–6 d of disease onset (1). Next-generation sequencing (NGS) enables the rapid recovery of whole genome sequences directly from clinical samples (2). NGS has been critical in determining the geographic lineage, infectious origin, and transmission chain of emerging viral outbreaks. Currently, long DNA or RNA fragments can be sequenced in real-time using portable and flexible nanopore sequencing. Numerous studies have used nanopore sequencing to help diagnose viral infections and characterize genomes (3, 4, 5, 6). Furthermore, CHIKV genotypes have been successfully determined using amplicon-based NGS targeting the envelope gene (7).
The first case of CHIKV infection was reported in 2013 in the Republic of Korea (ROK) (8). Although whole-genome analyses of the virus have not been performed in the country, 10 or more cases of CHIKV infection are reported annually since 2022 in travelers returning from outbreak areas, particularly Southeast Asia (9).
This study is the first to sequence the entire CHIKV genome from the serum sample of a Korean traveler returning from an endemic area. Our findings highlight the need for surveillance studies on repatriates from endemic countries and further research on the epidemiology of CHIKV outbreaks worldwide.
MATERIALS AND METHODS
Ethical approval
The study protocol, involving collection of patient residual specimens, was approved by the Institutional Review Board (IRB) of Chuncheon Sacred Heart Hospital, Hallym University (IRB 2022-05-003). Written consent was obtained from participating patients.
RNA extraction and cDNA synthesis
Serum samples obtained 2 and 6 days after symptom onset from CHIKV infected patients were provided by Hallym Chuncheon Sacred Heart Hospital, Kangwon province, ROK. Total RNA was extracted using the TRIzol LS Reagent (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg total RNA using the High-Capacity RNA-to-cDNA kit (Applied Biosystems; Foster City, CA, USA) according to the manufacturer’s instructions.
Amplicon-based NGS
The cDNA was amplified using CHIKV-specific primer mixtures and the Solg 2X Uh-Taq PCR Smart mix (Solgent; Seoul, Republic of Korea) according to the manufacturer’s instructions. The first PCR cycling conditions were as follows: initial denaturation at 95˚C for 15 min; followed by 40 cycles of 95˚C for 20 s, 50˚C for 40 s, and 72˚C for 1 min; and a final elongation at 72˚C for 3 min. The second PCR assay was conducted in a 25 μL reaction mixture containing 12.5 μL 2X Uh-Taq PCR Smart mix, 1.0 μL of the first PCR product, 10.0 μL of 0.5 μM of each primer mixture (final concentration, 0.2 μM), and 1.5 μL distilled water. The sequencing library was prepared using a Ligation Sequencing Kit (SQK-LSK109) according to the standard protocols (Oxford Nanopores Technologies, UK) and then ligated to sequencing adapters. The purified library was sequenced using a MinION device by loading onto a FLO-MIN106 (R9.4.1; Oxford Nanopores Technologies, UK) flow cell.
NGS data analysis and phylogenetic analysis
Basecalling was performed using Guppy (v3.0.3) embedded in the MinKNOW system (Oxford Nanopore Technologies). The adaptor sequences were trimmed, and the filtered reads were assembled into a FASTQ file using Porechop (v.0.2.4). Viral reads were mapped to the reference genome sequence CHIKV Thail 2019 (Accession ID MN630017.1) and the consensus was extracted using Medaka (v.12.0).
Phylogenetic analysis
The genomic sequences of the CHIKV were aligned using the MUSCLE algorithm in MEGA 11. The best-fit substitution model was determined by its Bayesian Information Criterion (BIC) as GTR+G+I. Phylogenetic trees were generated using the maximum-likelihood method in MEGA 11. Topologies were assessed by bootstrap analysis of 1,000 iterations. Geneious Prime was used to align the amino acid sequence with the reference sequences for both non-structural and structural proteins utilizing Clustal Omega 1.2.2, followed by amino acid similarity analysis using the BLOSUM90 matrix.
Estimation of the zoonotic prediction potential of CHIKV
The viral genome was assessed and rated for its potential to cause zoonotic infections through the utilization of a machine learning model (10). This model determines the probability of human infection by analyzing the host range characteristics encoded in the viral genome. The features encompass biases in the arrangement of the viral genome, such as the proportional occurrence of each codon, amino acid, and dinucleotide. This particular method entailed delineating the viral genome in the FASTA format and manually ascertaining the open reading frames (ORFs). The ORFs in viral genome ORFs were predicted using a gene-finding program called Prodigal (11). Zoonotic potential rankings for the four genotypes of CHIKV were created by inputting files in fasta format, which included genome and metadata files, and specifying output file names. The zoonotic potential of the viruses was ranked using the machine learning model with a cut-off value of 0.293 as reported in a previous study (12). Zoonotic potential prediction probabilities were categorized by low, entire 95% confidence interval (CI) of predicted probability > cutoff value; medium, mean prediction > cutoff value; although the CI crosses it; high, mean prediction > cutoff value, with the CI crossing it; and very high, entire CI > cutoff value).
RESULTS
Case description of CHIKV from a patient with CF
A 48-year-old male traveler from Pattaya, Thailand, reported being bitten 30–40 times by mosquitoes prior to hospitalization. He presented to the emergency room with fever, chills, myalgia, and arthralgia. Physical examination revealed swelling in both hands, arthralgia in the second proximal interphalangeal joint on the right and the fourth distal interphalangeal joint on the left, and a maculopapular skin rash on the trunk. Given the location and persistent arthralgia, IgM antibody testing for CHIKV was requested 2 d after hospitalization. The patient’s fever, myalgia/arthralgia, and skin rash gradually improved over a 5-day hospitalization, resulting in his discharge. Four days after discharge, IgM antibody testing confirmed chikungunya fever (Table 1).
Table 1.
Laboratory diagnosis of a patient with Chikungunya fever
Phylogenetic analysis of CHIKV from a patient with CF
After 22 h of nanopore sequencing on a sample collected 2 d after hospitalization, nearly the entire CHIKV genome was covered, with a mean coverage of 99.2% and over 5,000× depth. The whole genome length was 11812 bp, consisting of nonstructural and structural open reading frame (ORF). The nonstructural ORF length was 7425 bp and the structural ORF length was 3747 bp. However, only 9.1% of the genome had a depth of over 9× coverage in a sample collected 6 d after hospitalization (Table 2). Phylogenetic analysis revealed that the CHIKV strain isolated from ROK in 2022 (Accession ID: OQ918703) clustered with the East Central South African lineage (Fig. 1). The amino acid similarity of CHIKV Korea 2022 showed significant relatedness to the ECSA genotypes, particularly to CHIKV THBKK19-15 (Accession ID: LC580264) isolated in Thailand in 2019, indicating 99.8% similarity for non-structural and 99.7% for structural amino acid sequences. The amino acid similarity was distant from other genotypes, ranging from 99.1% to 99.3% for the Indian Ocean, 95.8% to 96.8% for the Asian, and 94.5% to 95.6% for the West-African lineage (Fig. 2). These findings indicate a phylogenetic relationship between CHIKV circulation in Thailand and cases introduced to ROK via tourism. A total of 453 complete sequences of CHIKV were obtained from the NCBI database (Table 3).
Table 2.
NGS analysis results for patient sera with CHIKV on days 2 and 6
| Sample ID | Number of reads | Number of covered bases | Coverage rate (%) | Average depth |
|---|---|---|---|---|
| Korea2022_Day 2 | 340,449 | 11,719 | 99.21 | 5,588.98 |
| Korea2022_Day 6 | 1,695 | 784 | 9.09 | 9.09 |
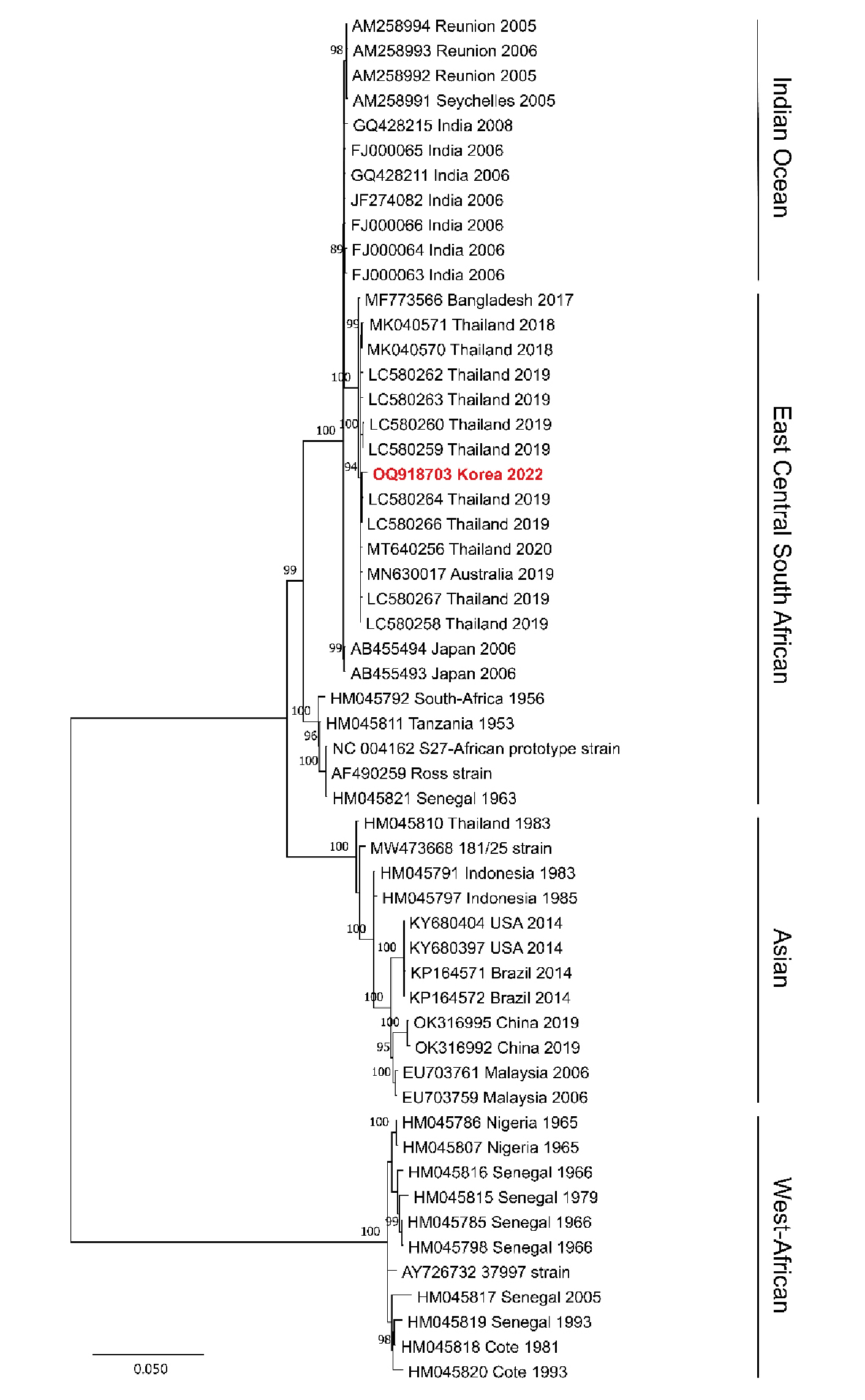
Fig. 1
Phylogenetic analysis of chikungunya virus (CHIKV) from a patient.
A phylogenetic tree was generated using the maximum likelihood method with the GTR model of the evolution and alignment of the CHIKV sequences. Colored lines show the phylogenetic lineage of CHIKV identified in East Central South African (blue), West African (orange), Indian Ocean (green), and Asian (yellow). The bold red arrow indicates the nearly whole genome sequence of CHIKV from a Korean patient who traveled to Thailand in this study. The topologies were evaluated using bootstrap analysis of 1,000 iterations. The numbers along the branches are bootstrap values. GenBank accession numbers are provided. Scale bar represents nucleotide substitutions per site.
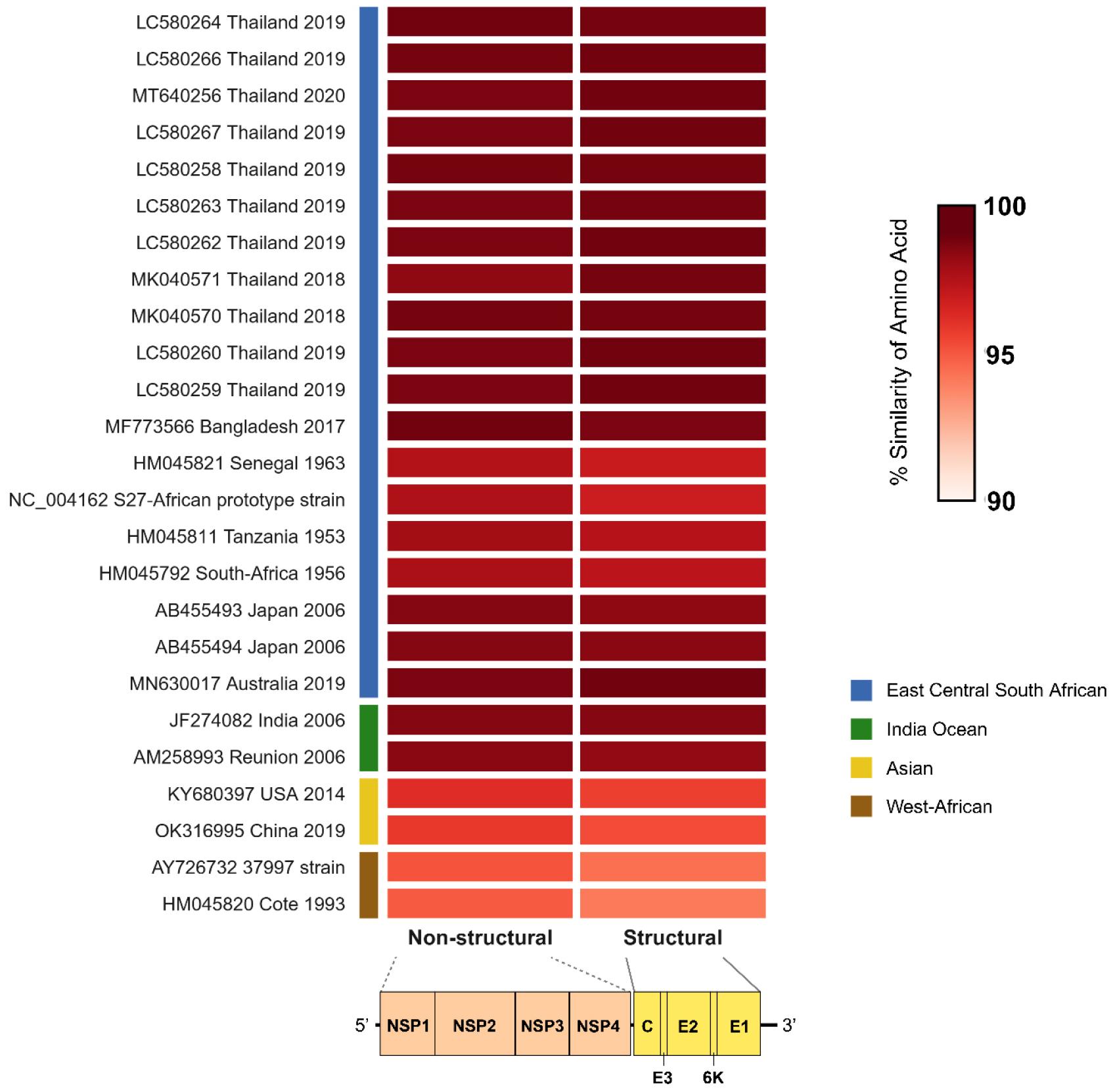
Fig. 2
Amino acid similarity of CHIKV Korea 2022 with ECSAs and other genotypes.
Heatmap illustrating the percentage similarity of Chikungunya virus (CHIKV) amino acid sequences used in this study compared with the East-Central-South African (ECSA) genotype and other CHIKV genotypes. The color gradient represents sequence similarity, with darker shades indicating higher similarity. Sequences are labeled with GenBank accession numbers, country of isolation, and year. Colored bars on the left denote different CHIKV lineages. The bottom panel categorizes two genomic regions encoding the precursors of non-structural (NSP1, NSP2, NSP3, and NSP4) and structural (C, E3, E2, 6K, and E1) proteins.
Table 3.
Characteristics of genomic sequences of chikungunya virus used in this study
Evaluating the zoonotic prediction potential of CHIKV by using the machine learning model
The zoonotic potential prediction score (median) of each genotype was 0.35 for WA, 0.52 for ECSA, 0.47 for IO, and 0.65 for Asian (Fig. 3). Notably, CHIKV Korea 2022 showed a zoonotic potential prediction score of 0.68. The zoonotic potential prediction presented that the WA genotype was medium, the ECSA and IO genotypes were high, and the Asian genotype was very high.
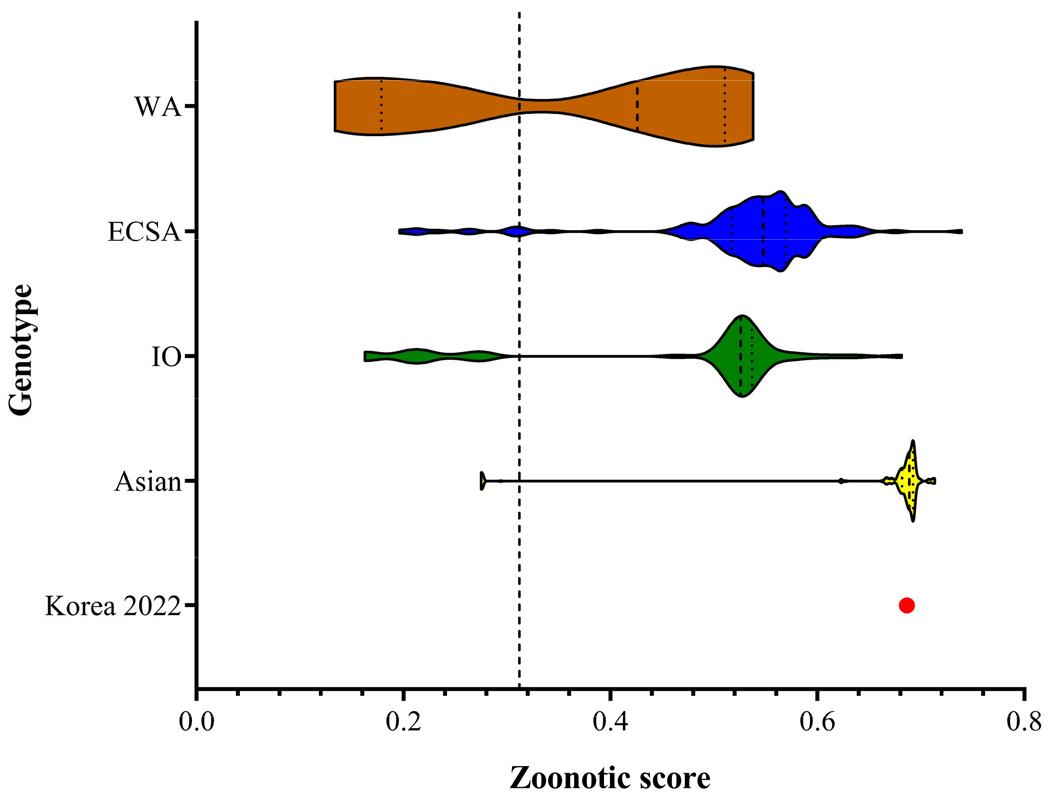
Fig. 3
Prediction of zoonotic potential for chikungunya virus (CHIKV).
Genotype-annotated CHIKV from NCBI GenBank were assessed for their zoonotic potential based on genome sequences. The distribution of zoonotic potential scores for CHIKV, stratified by genotype, is presented in a violin plot. Each genotype is distinguished by a specific color: WA (West African, brown), ECSA (East-Central Southern African, blue), IO (Indian Ocean, green), and Asian (yellow). The zoonotic potential score of CHIKV Korea 2022 identified in this study is designated by a red circle. The value of the cutoff for the optimal balance between sensitivity and specificity was 0.293.
DISCUSSION
The development and application of amplicon sequencing is crucial in enabling the differential diagnosis of CHIKV infections. This approach allows the rapid and targeted treatment of viral infections. The hallmark feature of nanopore sequencing, real-time sequencing, allows for faster full-length genome sequencing than other methods (10). To the best of our knowledge, this study is the first to show nearly complete whole-genome sequencing and genomic characterization of CHIKV in a Korean patient who had visited Thailand using amplicon-based nanopore sequencing. A clinical sample collected 2 d after hospitalization yielded nearly whole genome sequences of CHIKV, facilitating the examination of the patient’s CHIKV genotype. This study emphasizes the significance of prompt specimen acquisition in optimizing the efficiency of the amplicon-based NGS method when managing emerging infectious diseases.
Identification of the ECSA genotype in the patient who contracted the virus while visiting Pattaya, Thailand, confirms its presence in Asia. This finding is consistent with recent reports on ECSA genotype identification in Australian (13) and Japanese (14) locals and tourists. The availability of genomic data helps characterize the virus and establish a comprehensive database for monitoring the emergence of CHIKV strains worldwide. Active surveillance of mosquito-borne infectious diseases must be implemented for individuals returning from endemic areas (Fig. 4).
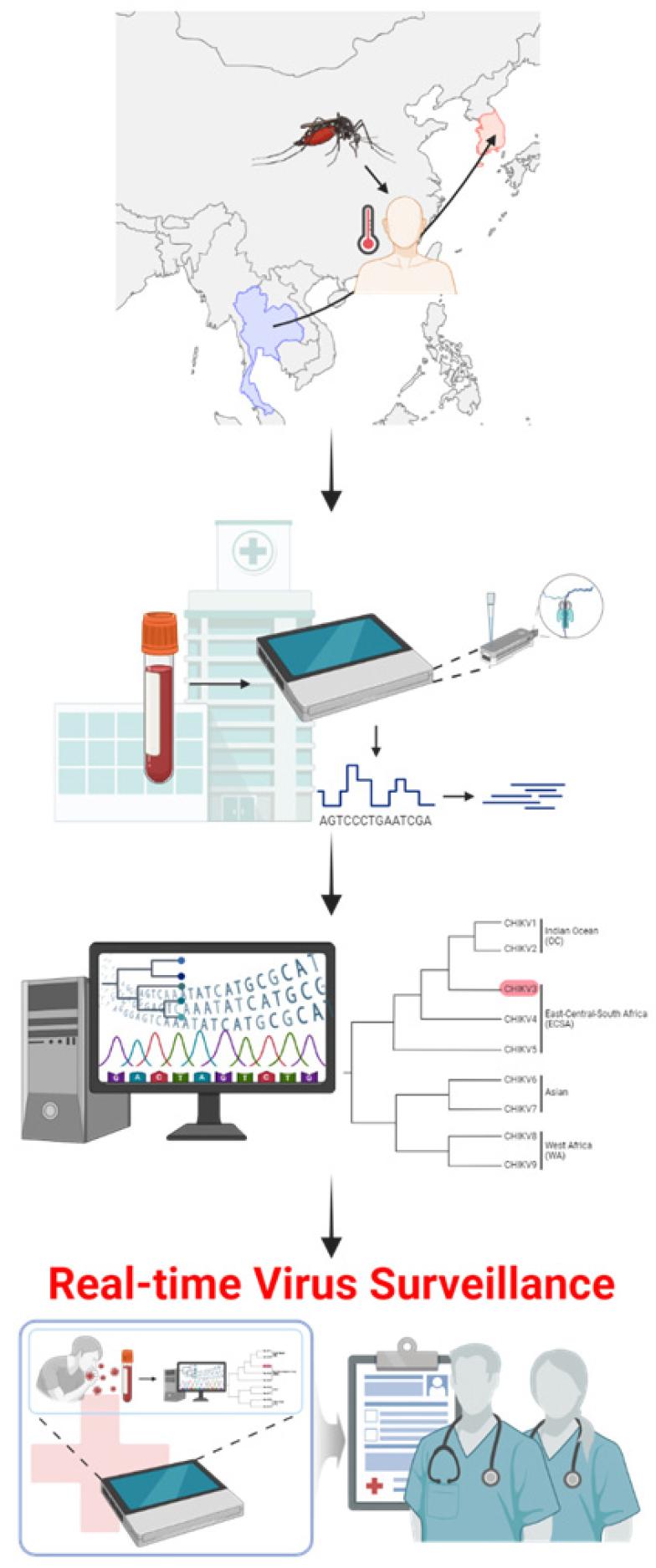
Fig. 4
An overview of real-time genomic surveillance for chikungunya virus (CHIKV) imported from endemic areas.Clinical samples from patients suspected of chikungunya fever, who travelled or worked in endemic regions, are collected at hospitals. Initial viral infection is confirmed by laboratory diagnosis, including RT-PCR and ELISA, from clinical specimens collected at the time of admission or within a few days thereafter. Amplicon-based MinION sequencing is employed to recover the genomic sequences of CHIKV, facilitating real-time on-site diagnosis of viral infection. Phylogenetic analysis rapidly delineates the suspected origin and source of etiological agents, revealing the genotype and geographical site of importation. Understanding the origin and transmission pathway of CHIKV is crucial for managing the patient influx in non-endemic regions and timely and effective medical responses. This real-time genomic surveillance arises awareness and caution for physicians regarding the introduction of the arthropod-borne virus from endemic to non-endemic areas. The illustration was created using BioRender.com.
Southeast Asia, which has a mosquito-friendly climate, remains a top travel destination worldwide (15). Consequently, CHIKV infection cases among tourists returning from Southeast Asia to their home countries continue to be reported, resulting in local transmission and increasing concerns about CHIKV infections in non-endemic regions (16). Rapid differential diagnosis of CHIKV infection is critical in preventing further transmission, as local transmission occurs even in previously non-endemic countries, as observed in Italy in 2007 (17). Consequently, prioritizing preparedness for emerging CHIKV infections is imperative regardless of geographical or climatic disparities.
Recently, zoonotic virus outbreaks become a significant human threat for public health (18, 19). Defining genome sequences of the emerging virus is critical for assessing the zoonotic potential (20). Here, the zoonotic potential risk of CHIKV demonstrated that the first discovered WA genotype was considered “medium” with the ZPS of 0.35 (21). However, the Asian genotype that circulated in the American continent in the 2010s was considered “high” with the ZPS of 0.65 (22). Notably, CHIKV Korea 2022 showed the ZPS of 0.68, leading to the hypothesis that CHIKV is very likely to evolve to adapt in humans over time (23, 24, 25). The ZPS was built using a machine learning model based on viral genome composition associated with human infection, and our analysis showed an increase in ZPS from older to more recent CHIKV genotypes. This could reflect some level of adaptation of CHIKV to the human host. Whether zoonotic potential prediction of CHIKV is associated with biological characteristics remains to be investigated in silico and in vivo.
This study has limitations owing to the small number of clinical samples. Consequently, further large-scale investigations on the specificity and sensitivity of the assay using additional clinical samples are warranted. Further studies should aim to develop a universal CHIKV primer pool capable of detecting all four genotypes.
In conclusion, amplicon-based MinION sequencing elicited the nearly whole genome sequence of CHIKV in a Korean patient with chikungunya fever, aiding in the definition of genotype and characteristics imported from the endemic area. This report provides significant insight into public health and travel medicine for effective strategies to mitigate the emergence of CHIKV in endemic and non-endemic regions.
