

INTRODUCTION
Viruses are a pervasive component of the human holobiont; even asymptomatic hosts harbor rich viral communities whose interactions do not always end with the death of the virus-infected cells (1, 2, 3). Recognizing the virome as a significant facet of human biology has sharpened interest in profiling its taxonomic and phylogenetic structure to elucidate roles in health and complex disease and to inform new diagnostic and therapeutic avenues (2, 4). Classical virus discovery via isolation and culture is slow and often infeasible because many viruses are recalcitrant to cultivation (5). Unlike bacteria, viral communities are also hard to characterize since there is no common gene for all viral genomes, preventing the application of ribosomal DNA profiling commonly used for bacteria studies. Consequently, unbiased shotgun metagenomics has become the primary route for virome characterization, enabled by next-generation sequencing that delivers large data volumes at reduced cost (6).
Despite its promise, routine clinical adoption is limited by the need for standardized, validated wet-lab procedures and accreditation-compatible quality systems (7). A second barrier is computational issues. Billions of short reads must be assigned to highly diverse, rapidly evolving viral genomes. There are two major approaches to address computational challenge. First, assembly-based pipelines reconstruct contigs and then annotate them to reference databases (8, 9, 10). Constructing contigs yields longer coding regions that improve annotation and host/context inference at the cost of time, memory, and potential chimeras (11). Second approach is read-based profilers mapping which reads directly to reference genome databases, trading contiguity for speed and scalability (12, 13, 14).
Given these contrasting strategies and their distinct assumptions, benchmarking is essential to quantify how platform choice shapes virome inferences across body sites. In this study, we benchmarked three widely used read-based profilers - Kraken2, FastViromeExplorer, and ViromeScan - on whole-genome shotgun (WGS) datasets from human microbiome project (15). We compared read retention and contamination filtering, alpha and beta diversity, and taxonomic composition. We also summarized statistical separations between sites providing practical guidance for tool selection in human virome studies.
MATERIAL AND METHODS
Data retrieval and processing
Human microbiome project (PRJNA275349) data were downloaded from the European Nucleotide Archive database. Paired-end libraries were quality trimmed with Trimmomatic v0.36 and trimmed reads were mapped to the human genome (hg38) with Bowtie v2.5.4 to remove human derived sequences. Taxonomic profiling was conducted using ViromeScan (13), Kraken2 v2.1.3 (12) and FastViromeExplorer (14) to generate relative abundances of viral species identified in each sample.
Microbiome analysis was conducted by phyloseq and related packages in R software v4.3.1. To measure alpha diversities, Chao1 index and Shannon’s index method were used. Principal coordinate analysis (PCoA) of the Bray-Curtis distance was performed to determine the community structure using the vegan package. The Kruskal-Wallis test and the non-parametric permutation multivariate analysis of variance (PERMANOVA) tests were used to assess the statistical significances for alpha and beta diversities, respectively. To test differential abundance of viral species among groups, linear discriminant analysis effect size (LEfSe) (16) was applied with default settings.
RESULTS
Read counts during Preprocessing
Raw WGS reads from four body sites were quality/adapter-trimmed with Trimmomatic and then mapped to the human genome with Bowtie to remove host reads. Trimming retained 86.0 ± 10.3% of gut reads, 11.5 ± 12.3% of nasal reads, 52.5 ± 25.0% of oral reads, and 7.1 ± 2.4% of vaginal reads. After human decontamination, the proportion of reads retained (Bowtie/Trim) was 100.0% (gut), 67.7 ± 19.4% (nasal), 95.0 ± 8.7% (oral), and 52.7 ± 17.9% (vaginal) (Table 1). These results indicate minimal human contamination in gut, moderate human contamination in oral datasets, and substantially higher host content in nasal and vaginal datasets.
Table 1.
Summary of data preprocessing
Unmapped reads were profiled for viral content using each platform and normalized to counts-per-million (CPM). Across sites, FastViromeExplorer generally reported the highest viral CPM, ViromeScan the lowest, and Kraken2 was intermediate. Overall, FastViromeExplorer yielded substantially higher viral signal in nasal, oral, and vaginal samples, while ViromeScan reported markedly lower CPM in all sites. Gut results showed no difference between Kraken2 and FastViromeExplorer but both exceeded ViromeScan (Table 2). Thus, choice of the platform strongly affected total viral profiles.
Table 2.
Virus counts depending on platforms
Microbial Diversity Differences depending on Platforms
To evaluate differences in viral diversity between platforms, the alpha diversity was assessed using the Chao1 and Shannon indices. Within-sample viral diversity differed markedly depending on platforms. FastViromeExplorer yielded the highest Chao1 and Shannon indices in most sites (nasal, oral, vaginal), Kraken2 was intermediate, and ViromeScan was consistently lowest (Fig. 1). In gut, Kraken2 and FastViromeExplorer were comparable, both exceeding ViromeScan. Across sites, Chao1 patterns varied by platforms. Vaginal samples had the lowest richness with Kraken2 and FastViromeExplorer, whereas the gut was lowest with ViromeScan. Nasal had the highest Chao1 in all platforms. Overall, platforms strongly influenced alpha diversity estimates, mirroring CPM trends.
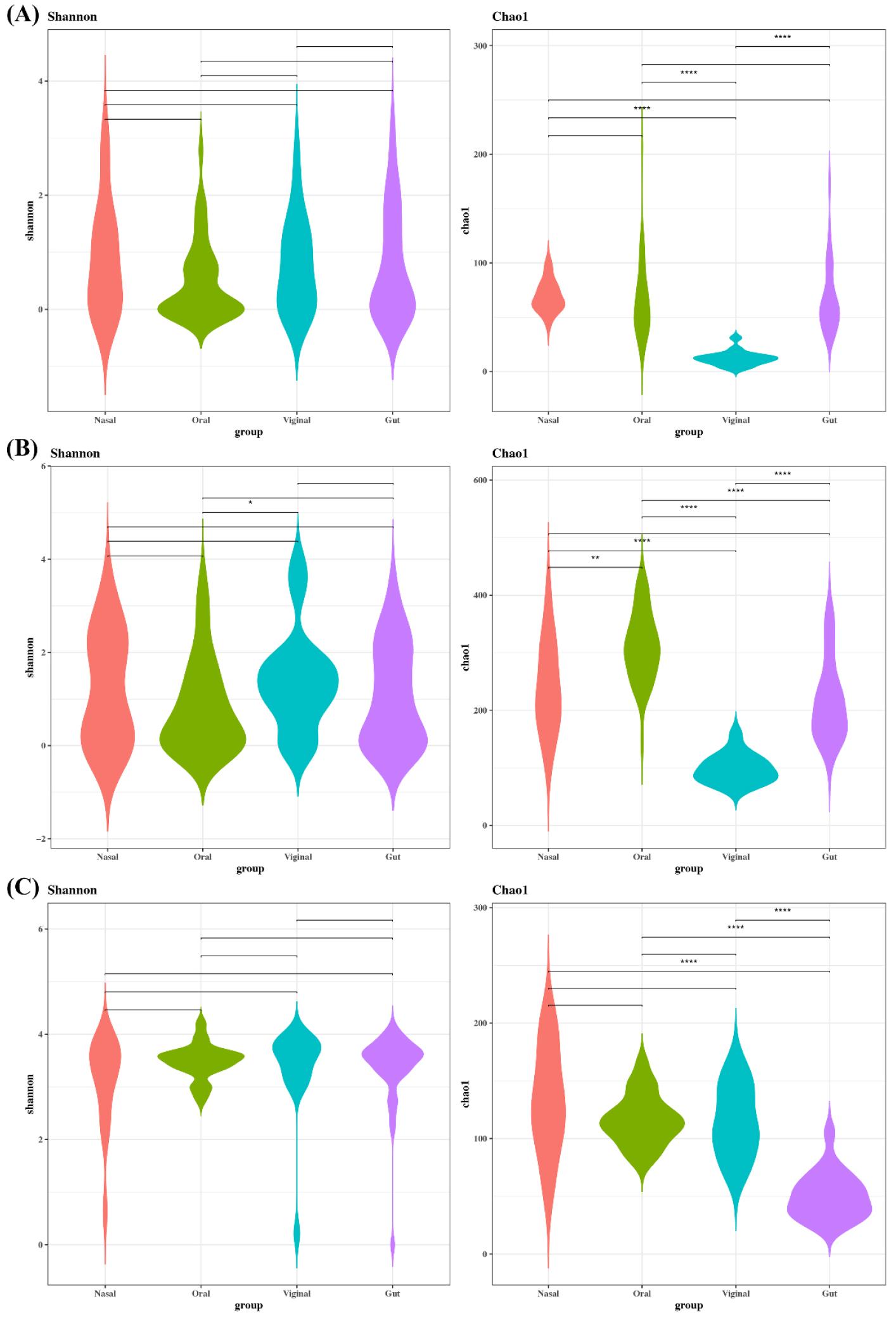
Fig. 1
Alpha diversity of sampling site depending on platforms. (A) Kraken2, (B) FastViromeExplorer, and (C) ViromeScan. Alpha diversity was used to describe the microbial richness and evenness within samples using the Chao1 and Shannon index.
Beta diversity was analyzed depending on platforms to determine the community composition. Ordinations revealed clear site-wise clustering with FastViromeExplorer showing the strongest separation among sites, Kraken2 showing moderate separation, and ViromeScan the weakest, partially overlapping clusters. Kraken2 performed all pairs significant after adjustment (Fig. 2). FastViromeExplorer performed all pairs significant except nasal–vaginal which was borderline. ViromeScan performed all pairs significant except nasal–vaginal. Effects comparable with the largest for gut–vaginal. Overall, site explains a substantial share of community differences, but the magnitude of separation depended on the platforms, while nasal–vaginal showed consistently the weakest contrast.
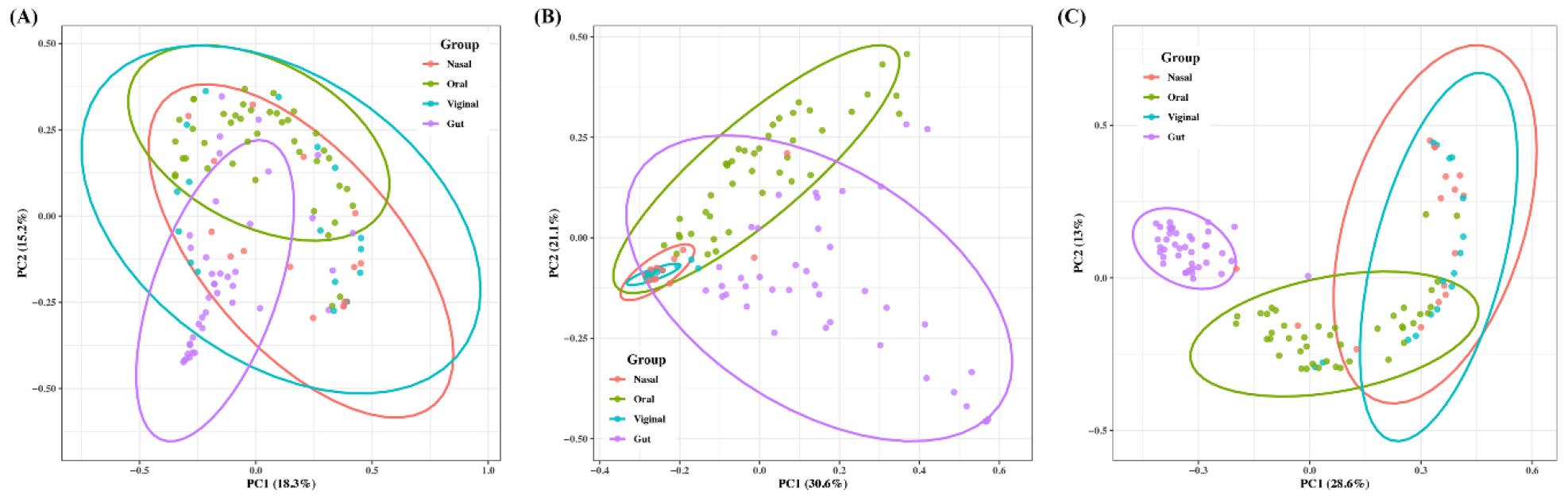
Taxonomic Composition at the Phylum Level Across Platforms
With Kraken2, most sites were dominated by Uroviricota. With FastViromeExplorer, Uroviricota was also dominant in nasal and vaginal, while oral and gut showed notable contributions from Lenarviricota, Negarnaviricota, and Cossaviricota. With ViromeScan, Uroviricota was not profiled, and communities were led by Nucleocytoviricota, followed by Peploviricota, Artverviricota, Cossaviricota, and Negarnaviricota. By site, gut and oral profiles were concordant between Kraken2 and FastViromeExplorer, whereas nasal and vaginal showed greater divergence (Fig. 3). Overall, platform choice materially changes which phyla appear among the phyla and their relative weights.
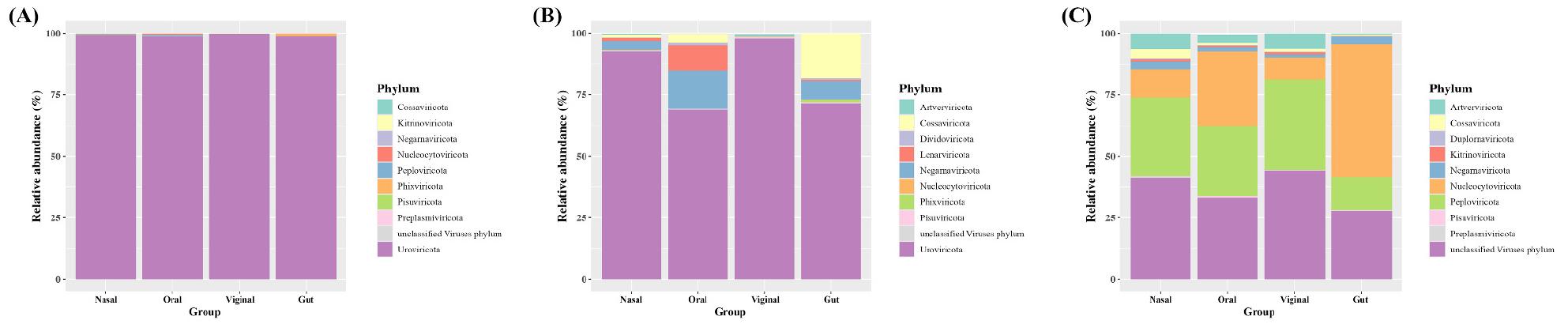
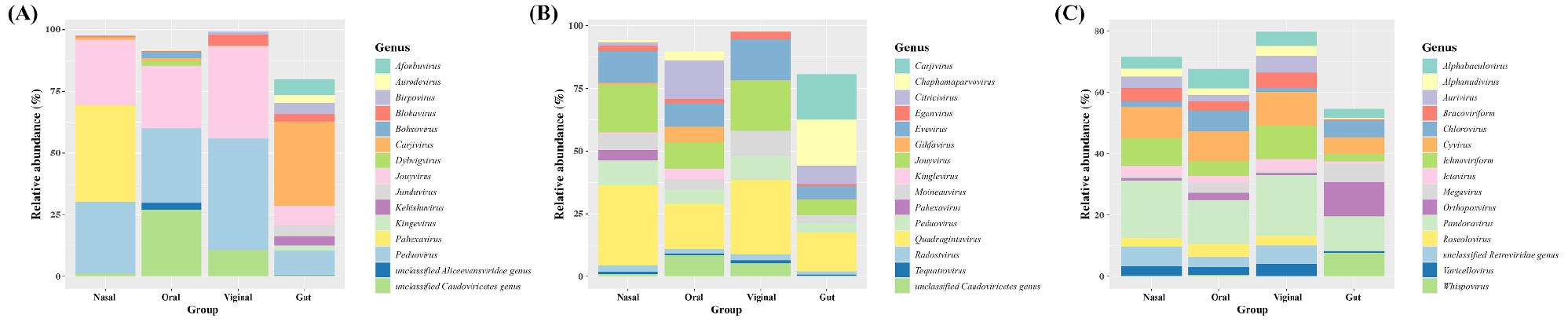
Taxonomic Composition at the Genus Level Across Platforms
At the genus-level, a total of 481 genera were identified through Kraken2, 855 genera through FastViromeExplorer and 108 genera through ViromeScan. Among these, 321 genera were commonly detected between Kraken2 and FastViromeExplorer, while 92 genera were commonly found between FastViromeExplorer and ViromeScan and 63 genera between Kraken2 and ViromeScan.
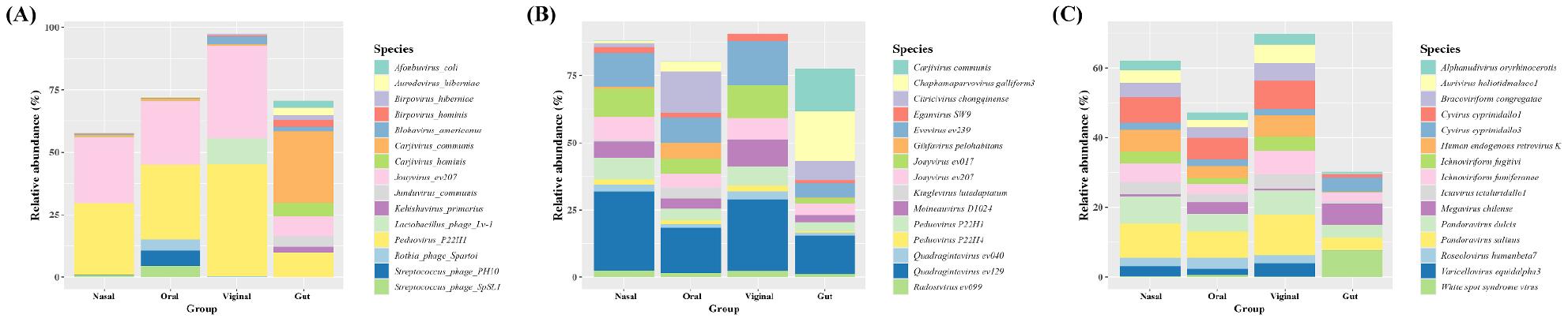
With Kraken2, Peduovirus and Jouyvirus were commonly found to be one of the most abundant across all sites with several site-distinctive genera. In the gut, Carjivirus was most abundant, followed by Peduovirus, Jouyvirus, Afonbuvirus, and Birpovirus. In nasal, Pahexavirus was most abundant. In oral samples, Peduovirus dominated alongside an unclassified Caudoviricetes genus and Jouyvirus. In vaginal samples, Peduovirus and Jouyvirus were dominant, with unclassified Caudoviricetes, Blohavirus and Birpovirus (Fig. 4A).
With FastViromeExplorer, Caudoviricetes-associated genera including Quadragintavirus, Peduovirus, and Jouyvirus were detected across all sites. In gut, top 5 most abundant genera were led by Chaphamaparvovirus, Carjivirus, Quadragintavirus, Citricivirus, and Jouyvirus. In nasal samples, Quadragintavirus and Jouyvirus were most abundant followed by Evevirus, Peduovirus, and Moineauvirus. In oral samples, Quadragintavirus and Citricivirus were most abundant, followed by Jouyvirus and Evevirus. In vaginal samples, the profile resembled nasal and oral samples, with Quadragintavirus and Jouyvirus most abundant, followed by Evevirus, Moineauvirus, and Peduovirus (Fig. 4B).
With ViromeScan, giant-virus including Pandoravirus, Orthopoxvirus, and Megavirus and parasitoid-associated groups including Ichnoviriform and Bracoviriform were frequently noted. In gut samples, the top 5 genera were Orthopoxvirus, Pandoravirus, Whispovirus, Megavirus, and Cyvirus. Pandoravirus and Cyvirus were commonly most abundant genera in nasal, oral, and vaginal samples. In nasal samples, Ichnoviriform, unclassified Retroviridae, and Bracoviriform were also abundant. In oral samples, Chlorovirus, Alphabaculovirus, and Ichnoviriform were also abundant. In vaginal samples, Ichnoviriform, unclassified Retroviridae, and Aurivirus were also abundant (Fig. 4C). These contrasts underscored strong platform effects on genus-level calls and suggest that conclusions about site-enriched genera are method-dependent; when biologically critical, key taxa should be validated across tools and reference databases.
Taxonomic Composition at the Species Level Across Platforms
At the species level, Kraken2 detected 982 species, FastViromeExplorer detected 2,272 species, and ViromeScan detected 254 species. Pairwise overlaps were limited; 1 species was common between Kraken2 and FastViromeExplorer, 182 species were common between FastViromeExplorer and ViromeScan, and there were no common species between Kraken2 and ViromeScan.
With Kraken2, Peduovirus P22H1 and Jouyvirus ev207 were recurrently most abundant across sites. In gut samples, Carjivirus communis was most abundant followed by Peduovirus P22H1, Jouyvirus ev207, Carjivirus hominis, and Junduvirus communis. In nasal samples, Pahexavirus P105, PHL082M03, PHL041M10 were notable nasal-enriched species. In oral samples, Streptococcus phage PH10, Streptococcus phage SpSL1, and Rothia phage Spartoi were also prominent. In vaginal samples, Lactobacillus phage Lv-1, Blohavirus americanus, and Blohavirus faecalis were also among the top 5 species (Fig. 5A).
With FastViromeExplorer, Caudoviricetes-linked species including Quadragintavirus, Peduovirus, Evevirus and Jouyvirus were widespread with site-specific highlights. In gut samples, Chaphamaparvovirus galliform3, and Quadragintavirus ev129, were also in the list. In oral samples, Citricivirus chongqinense, and Gihfavirus pelohabitans were also noted (Fig. 5B).
With ViromeScan, giant/parasitoid-virus groups were dominant species. In gut samples, Orthopoxvirus, Pandoravirus salinus/dulcis, Whispovirus, Megavirus chilense, and Cyvirus cyprinidallo1 were top 5 species. In nasal, oral and vaginal samples, Pandoravirus dulcis, Cyvirus cyprinidallo1, and Human endogenous retrovirus K were among the top 5 abundant species (Fig. 5C). Taken together, species-level calls are strongly platform-dependent in both coverage (detected set size) and composition (top species by site).
Platform-Dependent Differences in Microbial Profiles
Finally. LEfSe identified platform-specific species biomarkers (Fig. 6). Kraken2 yielded the largest set of significant species, while FastViromeExplorer yielded the fewest. With Kraken2 (Fig. 6A), multiple Streptococcus phages were enriched in oral samples, and several Pahexavirus species were enriched in nasal samples. With FastViromeExplorer (Fig. 6B), Streptococcus phages were again oral-enriched, whereas Lactobacillus phages were characteristic of vaginal samples. With ViromeScan (Fig. 6C), Gammapapillomaviruses were prominent in nasal samples; in oral samples, Varicellovirus, Simplexvirus, and Omegapapillomavirus were significant. In vaginal samples, Alphapapillomavirus, Simplexvirus, Molluscum contagiosum virus, Cytomegalovirus, and Varicellovirus were among the significant species. Taken together, the biomarker set varies strongly by platform, mirroring database coverage and mapping strategy.
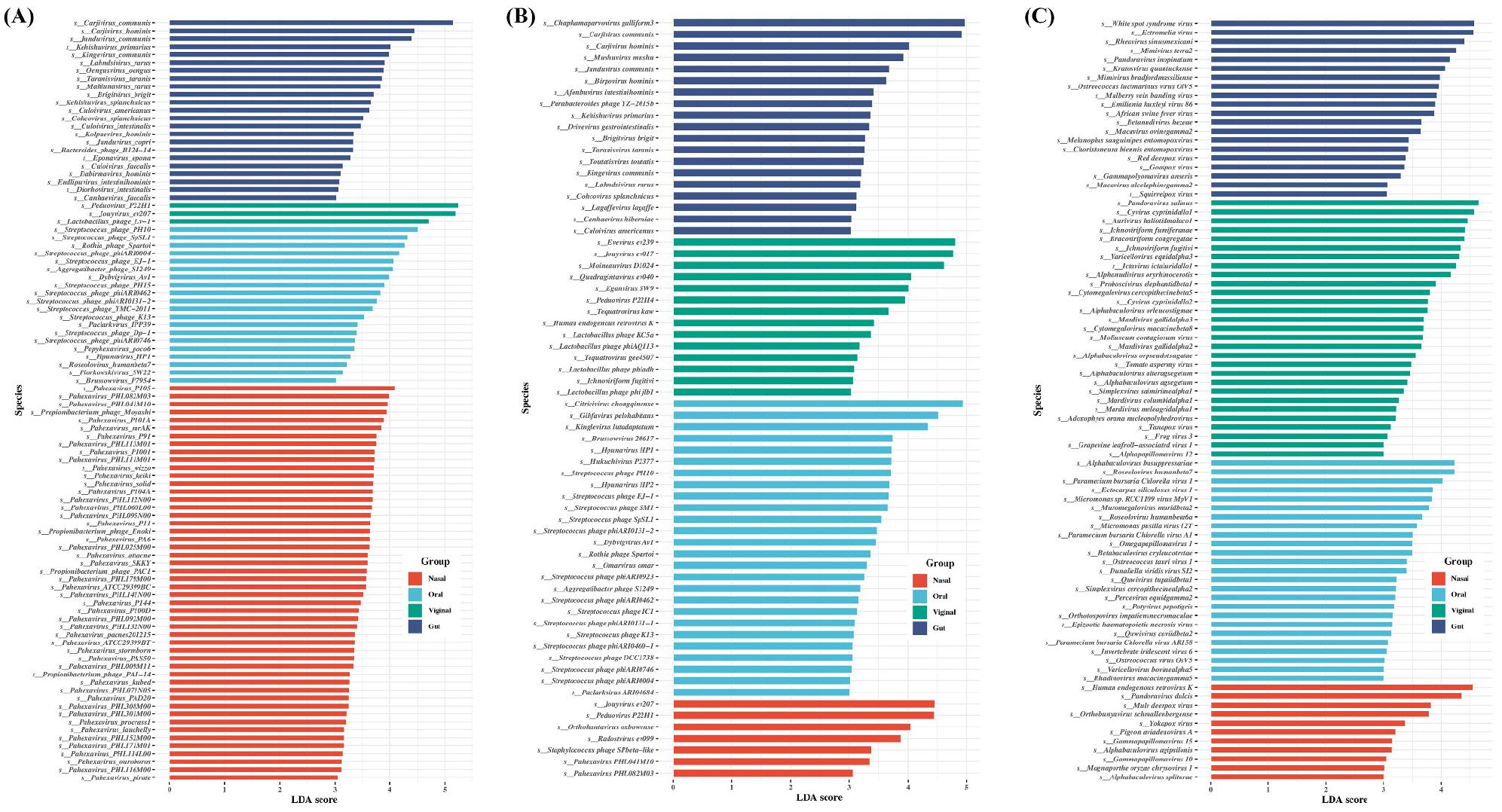
Fig. 6
Comparisons of the viral abundance depending on sampling sites showing significant differences. (A) Kraken2, (B) FastViromeExplorer, and (C) ViromeScan. The analysis was performed using linear discriminant analysis (LDA) and effect size analysis (LEfSe). Significant viral species with an LDA score >3.0 at species level were plotted.
DISCUSSION
Human virome is a pervasive, biologically consequential component of the holobiont. However, it remains hard to measure because viruses lack a universal marker and many are uncultivable. Shotgun metagenomics has become the primary route for virome characterization, but clinical translation is constrained by the computational challenge of assigning billions of reads. There are two major analytic approaches; assembly-based pipelines and read-based profilers. Assembly-based pipelines construct contigs that improve annotation and host/context inference. However, there are several challenges and limitations of contig assembly (11). High-abundance taxa assemble well while rare members fragment, biasing downstream diversity and function estimates (17). Large, complex metagenomes require substantial RAM and CPU and results vary with assembler, k-mer choices and versions (18). Mapping back to assembled contigs can shift abundance estimates relative to direct read-profiling, complicating cross-study comparisons (19). Thus, we benchmarked read-based profilers to understand how tool choice alters inferred diversity and composition across body sites.
Across four body sites, read-based profilers produced markedly different viral signals. FastViromeExplorer consistently yielded higher CPM and greater alpha-diversity than Kraken2, with ViromeScan lowest overall. There are several factors for these discrepancies. First, reference database design could be one of the factors. Kraken2 and FastViromeExplorer rely heavily on phage/RefSeq-centric catalogs, whereas ViromeScan emphasizes curated human and eukaryotic-virus genomes and applies hierarchical filters. A database weighted to bacteriophages favors overall virome discovery, while a human-virus-focused catalog prioritizes clinically relevant viruses but may under-call phages and novel lineages. Second, mapping strategy and stringency could be another reason. Kraken2 uses exact k-mer matches with lowest common ancestor (LCA) assignment (12), while FastViromeExplorer uses pseudoalignment (kallisto) plus coverage, ratio, and read filters (14) and ViromeScan uses staged mapping after host and bacterial pre-filters (13). These choices could shift sensitivity–specificity trade-offs. Third, site biology and host carryover could have influenced the profiles. Nasal and vaginal libraries carried more host reads and lower retained fractions after decontamination, amplifying differences among tools. Viral communities are also niche-specific and database emphasis may have interacted with site biology.
Our cross-platform comparison shows that database scope and matching strategy reshape the virome picture. At the phylum level, Kraken2 and FastViromeExplorer yielded Uroviricota dominance in most sites. Uroviricota is a phylum of non-enveloped dsDNA viruses that includes the class Caudoviricetes, which is well known to have a distinct shape. The virion has an icosahedral head that contains the viral genome and is attached to a flexible tail by a connector protein (20). ViromeScan reported communities led by Nucleocytoviricota, followed by Peploviricota, Artverviricota, Cossaviricota, and Negarnaviricota. Nucleocytoviricota is a large dsDNA viruse including family of Poxviridae and Pandoraviridae(21, 22). These viruses are referred to as nucleocytoplasmic since most of the viruses in this family replicate in both the host’s nucleus and cytoplasm. Peploviricota is a dsDNA virus that includes order of Herpesvirales, characterised by a common morphology consisting of an icosahedral capsid enclosed in a glycoprotein-containing lipid envelope. Families of Herpesvirales include Alloherpesviridae, Malacoherpesviridae, and Orthoherpesviridae (23). Cossaviricota is a phylum of viruses, whose named after Yvonne Cossart who discovered Parvovirus B19, the causative pathogen of fifth disease (24). Cossaviricota include class of Mouviricetes, Papovaviricetes, and Quintoviricetes(25).
LEfSe highlighted platform-specific viral biomarkers when each site was contrasted against the others, reinforcing that database scope and mapping strategy shape biological conclusions. Kraken2 produced the largest biomarker set and ViromeScan emphasized eukaryotic viruses, consistent with their reference emphases. In oral samples profiled by Kraken2 and FastViromeExplorer, repeated enrichment of Streptococcus phages aligns with the Streptococcus-rich oral microbiome and frequent oral phage activity. In nasal profiled with Kraken2, Pahexavirus fit a phage-leaning nasal community, while ViromeScan profiling noted Gammapapillomaviruses, plausibly reflecting mucosal epithelia of the anterior nares. In vaginal samples profiled by FastViromeExplorer, Lactobacillus phages were coherent with a Lactobacillus-dominated niche, suggesting active phage–host dynamics.
Taken together, distinct goals may require different analytical strategies. For human virus detection such as clinical or targeted use, the primary object is to detect sensitive but high-specificity pathogenic or clinically actionable human viruses (e.g., herpesviruses, papillomaviruses), with interpretable evidence at the genome level. In this study, ViromeScan profiled many eukaryotic and giant-virus calls and human endogenous retrovirus hits, while FastViromeExplorer and Kraken2 mainly emphasized phages. For clinical questions, a human and eukaryotic-virus-prioritized database and stringent filters should be preferable. For overall virome discovery such as ecology or evolution study, the primary object is a comprehensive cataloging of viral diversity, especially phages and novel lineages, inference of community structure, dynamics, and putative functions. FastViromeExplorer and Kraken2 produced higher richness and stronger between-site structure, which is useful for ecological contrasts. On the other hand, ViromeScan under-called phages and compressed beta-diversity, limiting discovery power. Platform-dependent phylum or genus shifts show that database breadth steers ecological narratives.
There are several limitations and caveats. Read-based profilers cannot recover novel viruses absent from the reference. Databases evolve rapidly and our conclusions reflect specific versions and may shift as catalogs expand. Host depletion and library prep differ across sites and residual host reads can suppress viral detection and bias tool comparisons (26).
CONCLUSION
Human virus detection and overall virome discovery are related but different problems. The former prioritizes specificity, interpretability, and confirmability, wihle the latter prioritizes breadth, sensitivity, and novelty capture. Our benchmark shows that platform choice materially alters both the amount of viral signal and the story one tells about virome composition. Choosing a pipeline aligned to the biological question is therefore essential.
