

INTRODUCTION
The coronavirus disease 2019 (COVID-19) is caused by a newly emerged virus, named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1, 2). Since its first reporting on December 2019 in Hubei Province, China, COVID-19 has rapidly spread globally, and the World Health Organization (WHO) declared it a pandemic on March 11, 2020 (3). As of April 20, 2020, 221,134,742 human confirmed cases of COVID-19, including 4,574,089 deaths, have been reported from 213 countries (4). The first case in South Korea, a Chinese visitor from Wuhan, was identified on January 20, 2020 (5, 6). On February 3, 2020, the first case in the city of Gwangju, South Korea, was reported, and as of September 2021, a total of 4,364 confirmed cases have been identified.
SARS-CoV-2 is an enveloped, positive-sense single-stranded RNA virus (7). The SARS-CoV-2 genome encodes 16 non-structural proteins (nsps) involved in virus replication and four structural proteins, including envelope (E), membrane (M), and nucleocapsid (N), and spike (S) (8). RNA viruses have higher mutation rates than DNA viruses (9). Mutations in the SARS-CoV-2 genome are being continuously reported (10, 11), and these mutations may affect pathogenesis. Therefore, it is critically important to monitor the evolution of SARS-CoV-2. Investigation of the viral genomic variations is necessary to provide epidemiological information on SARS-CoV-2 and for the development of therapeutics and vaccines.
In this study, we isolated SARS-CoV-2 virus from COVID-19 patients in the city of Gwangju, South Korea, and we investigated genomic mutations resulting in amino acid changes. To this end, we performed full-genome sequencing using a next-generation sequencing (NGS) tool and analyzed point mutations in the SARS-CoV-2 genome. We phylogenetically analyzed and classified virus strains from confirmed SARS-CoV-2-infected patients in the southwestern region of Korea.
MATERIALS AND METHODS
Sampling and viral RNA isolation
This study was approved by the Institutional Review Board (approval no. P01-202008-31-004) of Public Institutional Bioethics Committee designated by the Ministry of Health of Welfare (MOHW). Clinical specimens were collected as oropharyngeal and nasopharyngeal swabs in viral transport medium or sputum from symptomatic patients with COVID-19. All clinical samples were handled in biosafety cabinets. Viral RNA was extracted from the samples using a QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany). The RNA was quantified through the Korea Centers for Disease Control and Prevention (KCDC) method and using a PowerChek 2019-nCoV Real-time Kit (Kogene Biotech, Seoul, South Korea).
Real-time reverse-transcription polymerase chain reaction (RT-qPCR)
RT-qPCR assays targeted the RNA-dependent RNA polymerase (RdRp) and E genes. Sequence information for the primers and probes used to detect these two genes is presented in Table 1(12). PCRs were run in 25-μL reaction mixtures containing 12.5 μL of 2X RT-PCR buffer, 1 μL of 25X RT-PCR enzyme mixture included in AgPath-ID One-step RT-PCR Reagents (Thermo Fisher Scientific), 5 μL of RNA, and 1 μL of each primer/probe. The PowerChek 2019-nCoV Real-time PCR Kit includes a primer/probe mixture (for RdRp and E), a positive control template (for RdRp and E), and RT-qPCR premix. Sequences of the primers and probe are not disclosed. The RT-qPCR master mix consists of 11 μL of RT-qPCR premix, 4 μL of each primer/probe mixture, and 5 μL of RNA. The thermal cycling program was as follows: reverse transcription at 50°C for 30 min, followed by inactivation of the reverse transcription at 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min (7500 instrument; Applied Biosystems, Foster City, CA, USA). All samples were tested in duplicate.
Table 1.
RT-qPCR primers/probes (KCDC)
SARS-CoV-2 isolation in Vero cells
SARS-CoV-2 virus isolation and cultivation were all performed in the Biosafety level -3 Laboratory (BSL-3). To isolate SARS-CoV-2 from Vero cells, the cells were seeded in 24-well plates at 2×104 cells/well and cultured in modified Eagle’s medium (MEM; Gibco, Thermo Fisher Scientific, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco) for 48 h. Then, the medium was replaced with medium with a lower FBS concentration, and the cells were further incubated for 24–48 h. Before inoculation onto the Vero cells, patient specimens were diluted with viral transport medium containing penicillin-streptomycin (Gibco) at a 1:4 ratio, incubated at 4°C 1 h with vortexing every 15 min, and then centrifuged at 3,000 rpm for 30 min. Inoculated monolayers of Vero cells were incubated in MEM supplemented with 2% FBS at 37°C in the presence of 5% CO2. For seven days post inoculation the cells were observed for cytopathic effects (CPE) daily. When CPE was observed, the cells were scraped with a micropipette tip and subjected to total nucleic acid extraction.
Next-generation sequencing
Ten cell soup containing viral particles and six clinical samples with a threshold cycle (Ct) value of 10 or lower were used for genome analysis. Viral RNA was isolated using the QIAamp Viral RNA Mini Kit and subjected to whole-genome sequencing. NGS libraries were prepared and sequenced using Barcode-Tagged Sequencing (BTSeq) technology (Celemics Inc., Seoul, Korea) according to the manufacturer’s protocol. Complete genome sequencing was conducted using the Illumina MiSeq platform (Illumina, San Diego, CA), producing 150-bp pair-end reads per sample.
Phylogenetic analysis
Forty complete viral genome sequences were downloaded from the Global Initiative on Sharing All Influenza Data (GISAID; https://www.gisaid.org/) database, with acknowledgment. Multiple alignment of the SARS-CoV-2 genomes was carried out using Multiple Sequence Comparison by Log-Expectation (MUSCLE). A phylogenetic tree was generated by the neighbor-joining method using Molecular Evolutionary Genetics Analysis across Computing Platforms (MEGA X) using the Kimura 2-parameter model, and the tree was evaluated using 1,000 bootstrap replicates.
Mutation analysis
A reference SARS-CoV-2 genome (hCoV-19/Wuhan-Hu-1/2019) of 29,903 nucleotides long was obtained from the GISAID database and was used to identify mutations in the protein-coding sequences of the SARS-CoV-2 genomes of the Gwangju isolates. We analyzed 12 protein-coding sequences (ORF1a, ORF1b, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10). The amino acid sequence was created by using hCoV-19/Wuhan-Hu-1/2019 annotated open reading frames (ORFs). Based on the protein annotations, amino acid codon variants were converted from the nucleotide variants for alignments. Each protein domain was aligned with the reference genome of SARS-CoV-2 using NCBI protein alignments.
RESULTS
Virus isolation from positive samples
For virus isolation, SARS-CoV-2-positive clinical specimens with throat swab or sputum were inoculated into Vero cell cultures. We observed CPE at 24-h intervals for 7 days post inoculation. CPE was first observed and evident in Vero cells at 72–84 h after inoculation. When >80% of total cells showed CPE, the cells were collected (day 6), and viral RNA was extracted.
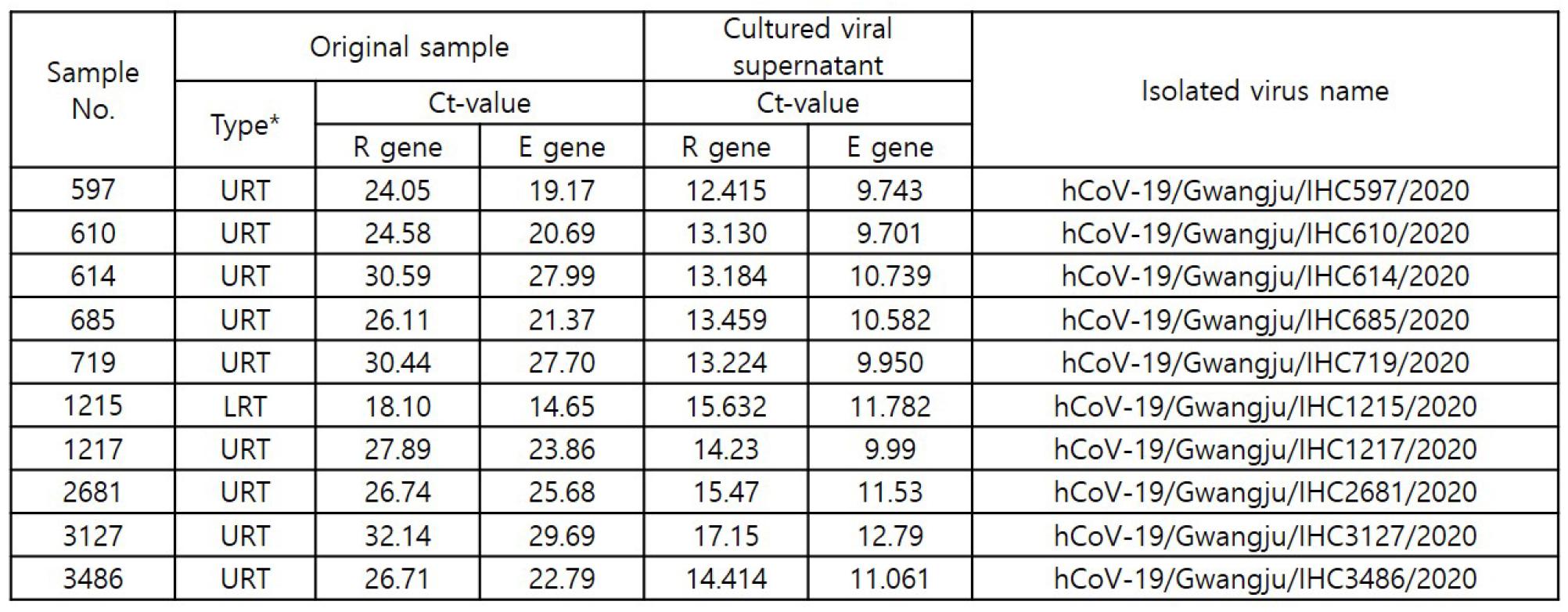
For the detection of SARS-CoV-2, the RdRp and E genes were detected using RT-qPCR. A positive RT-qPCR result was defined as a Ct value <37 for both genes. We successfully isolated SARS-CoV-2 from clinical samples collected from 10 COVID-19 patients in Gwangju. The virus isolates were named hCoV-19/Gwangju/IHC_sample number/2020 (Table 2).
Phylogenetic analysis of the SARS-CoV-2 genomes
We performed phylogenetic analyses of 16 distinct SARS-CoV-2 sequences of isolates from Gwangju and 40 sequences deposited in the GISAID database up to March 31, 2020 (Fig. 1). According to GISAID data, three major clades of SARS-CoV-2, including S, V, and G, can be identified (13). These three clades are determined by the amino acid substitutions present: ORF8-L84S (S), ORF3a-G251V (V), and S-D614G (G).
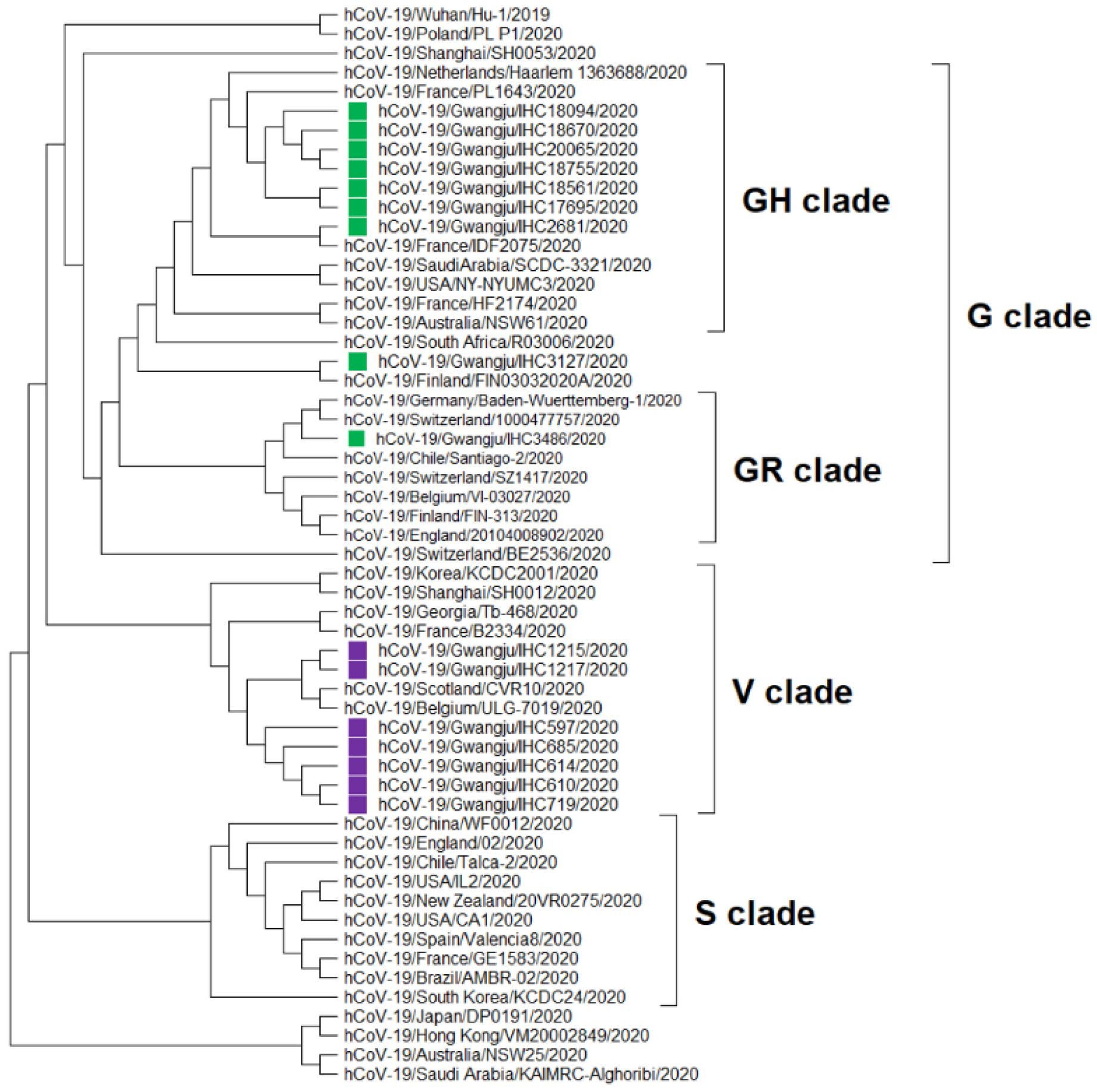
Fig. 1
Phylogenetic analysis of 56 representative SARS-CoV-2 genomes, including sequences of viruses collected in the city of Gwangju, South Korea (green and purple). Clades S, V, and G were determined based on significant amino acid mutations (ORF8-L84S, ORF3a-G251V, and S-D614G, respectively). The G clade was classified into a GR clade, a GH clade, and other strains. Multiple sequence alignment was achieved using MUSCLE. The phylogenetic tree was constructed by the neighbor-joining method using MEGA X and the Kimura 2-parameter model. The best tree was found by performing 1,000 bootstrap replicates.
In accordance with the above classification, we identified three clades (GH, GR, and V) on the basis of the point mutations identified in the Gwangju sequences (Fig. 1). Clade V included seven isolates from Gwangju. Five of these isolates (indicated in purple in Fig. 1, IHC597, 610, 614, 685, and 719) were associated with the Shincheonji religious group, and the remaining two (Fig. 1, IHC1215, and 1217) were related to another church in Gwangju. As revealed by the epidemiologic information, the five isolates from this clade were related to the spread during the early COVID-19 pandemic in South Korea. Specifically, in February 2020, the COVID-19 outbreak started in the city of Daegu, South Korea, and SARS-CoV-2 had rapidly spread within the religious group named the Shincheonji church of Jesus (14). The G clade comprised nine of the 16 isolates (labeled green in Fig. 1). We identified two subclades within clade G: GH, with seven isolates, and GR, with one isolate. Of the seven confirmed cases related to the seven GH isolates, six were linked to cluster secondary infections from door-to-door sales businesses at Geumyang Building (Fig. 1, IHC17695, 18094, 18561, 18670, 18755 and 20065). A Geumyang Building case led to a cluster outbreak in Gwangju from late June to mid-July and further led to additional cases at the Gwangneuksa temple, church, and work. The remaining one case appeared to be associated with international travel-related exposures (Fig. 1, IHC2681). The case related to the one isolate in clade GR had a known international travel history (Fig. 1, IHC3486).
Taken together, these results reveal that the V clade of SARS-CoV-2 was the major clade type in Gwangju during the early pandemic in South Korea, and as of mid-July in 2020, clade GH is the predominant type in the southwestern region of South Korea.
Mutations identified in the sequenced SARS-CoV-2 genomes
We aimed to classify the distinct clades of SARS-CoV-2 and investigate the mutations in the 16 SARS-CoV-2 genomes of the Gwangju isolates. To this end, we analyzed 12 protein-coding regions (ORF1a, ORF1b, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, and ORF10). NGS revealed 21 non-synonymous mutations and 1 frameshift deletion compared to the reference genome from a Wuhan isolate (NCBI GenBank: NC_045512). Mutation events observed in the sequenced SARS-CoV-2 genomes are summarized in Table 3.
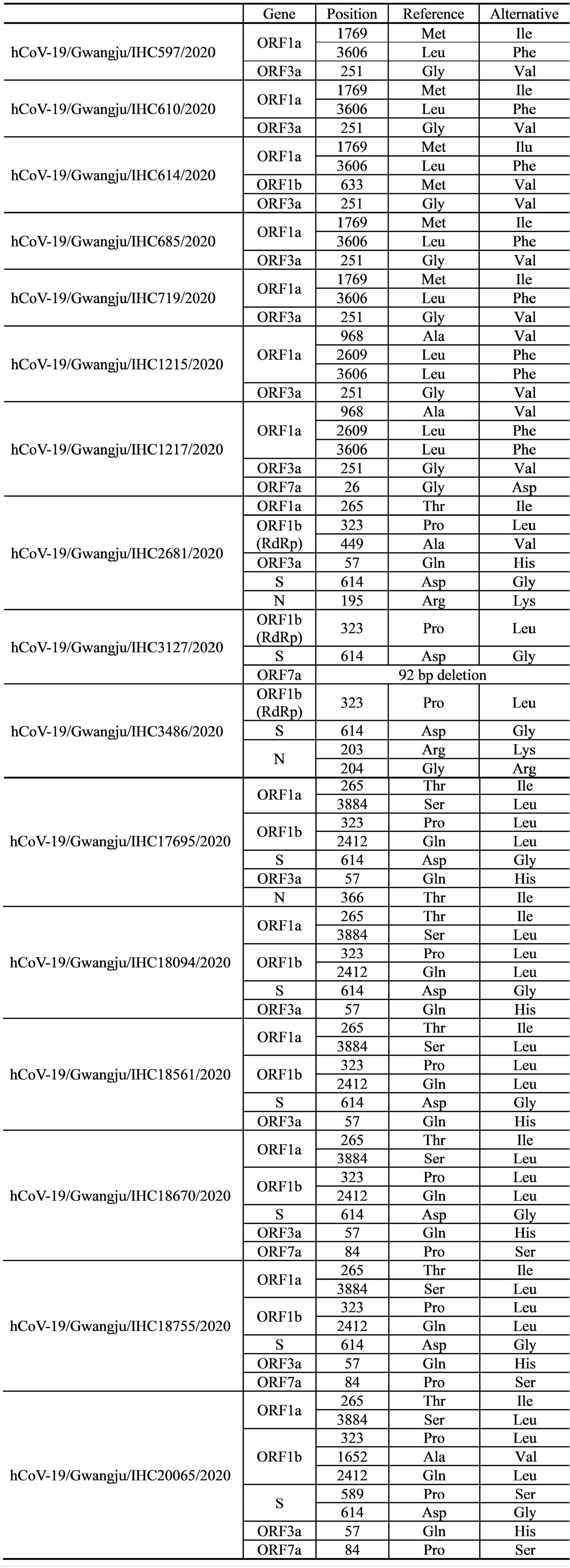
Clades V (ORF3a-G251V) and G (S-D614G) comprised seven and nine out of the 16 sequences of Gwangju isolates, respectively (Table 4). ORF8-L84S, representative of clade S, was not observed in any of the 16 genomes. In addition to the G251V mutation in ORF3a and the D614G mutation in S, we analyzed 19 additional point mutations in ORF1a, ORF1b, ORF3a, ORF7a, S, and N. For these 19 genetic mutations, 11 mutation sites were found in ORF1a and ORF1b, which encode nsp1–16, occupying two-thirds of the entire genome (Fig. 2).
Table 4.
Virus classification by representative mutations of each clade
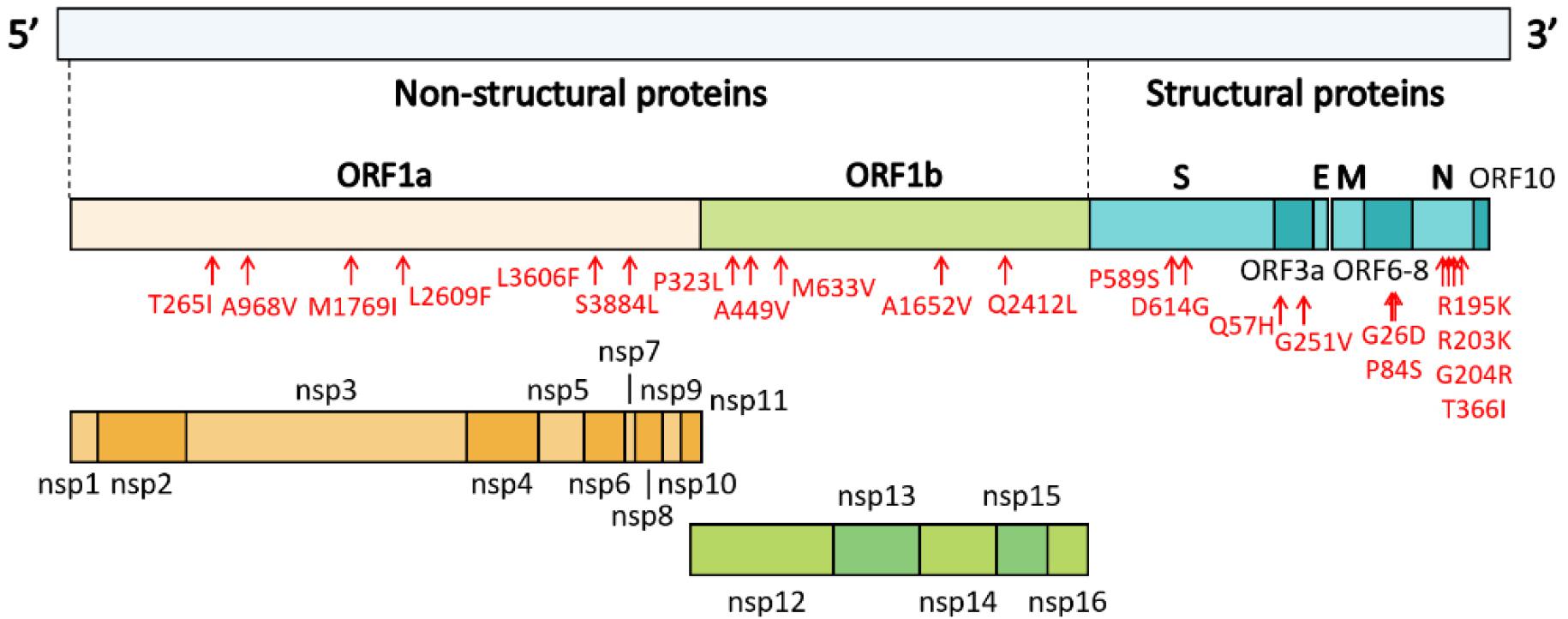
Fig. 2
Genome organization of SARS-CoV-2. ORF1a and ORF1b encode 16 non-structural proteins (nsp1-16). The structural genes encode four structural proteins, including spike (S), envelope (E), matrix (M), and nucleocapsid (N). Mutation sites identified in the sequenced genomes of isolates from Gwangju are indicated in red.
We analyzed the mutations in ORF1a/b, and we identified six mutation types in ORF1a (Table 3). Among them, the most common mutations were ORF1a-T265I (in seven samples, IHC2681, 17695, 18094, 18561, 18670, 18755, and 20065), ORF1a-L3606F (in seven samples, IHC597, 610, 614, 685, 719, 1215, and 1217) and ORF1a-S3884L (in six samples, IHC17695, 18094, 18561, 18670, 18755, and 20065). In particular, we found that ORF1a-L3606F co-occurred with ORF3a-G251V, which determines clade V (Table 3). Further, M1769I, A968V, and L2609F occurred in nsp3 within ORF1a. Mutations within nsp2 and nsp3 may play a role in differentiating infectivity of SARS-CoV-2 from SARS-CoV (15). In addition, we detected five mutations in ORF1b. P323L in nsp12 and Q2412L were observed in nine (IHC2681, 3127, 3486, 17695, 18094, 18561, 18670, 18755, and 20065) and six (IHC17695, 18094, 18561, 18670, 18755, and 20065) genome sequences, respectively. We also identified A449V and M633V in nsp12 and A1652V. The Nsp12 gene of SARS-CoV-2 encodes the RdRp protein essential for the virus replication machinery.
The remaining eight point mutations were found in ORF3a, ORF7a, S, and N. Four mutations, R195K, R203K, G204R, and T366I, were detected in the N protein, which participates in viral RNA genome packaging and viral particle release (16). Among them, G204R was observed together with ORF1b-P323L in the genome sequence of hCoV-19/Gwangju/IHC3486/2020 (Table 3). Based on GISAID data, two subclades, GR and GH, belonging to clade G, were determined by two mutations (N-G204R and ORF1b-P323L) and one mutation (ORF3a-Q57H), respectively. This indicates that isolate hCoV-19/ Gwangju/IHC3486/2020 can be classified into clade GR. Apart from the ORF3a-G251V and S-D614G substitutions, the single point mutations of Q57H in ORF3a and P589S in S were detected. In particular, ORF3a-Q57H, the signature of the GH clade, was detected in seven Gwangju genomes. In the ORF7a region, we confirmed two mutations; ORF7a-G26D and ORF7a-P84S.
In addition to the above 19 point mutations, we identified a 92-nucleotide deletion in ORF7a of hCoV-19/Gwangju/ IHC3127/2020, resulting in out-of-frame deletion. The SARS-CoV ORF7a ortholog is an inhibitor of bone marrow stromal antigen 2 (BST-2), which is capable of restricting virus release from cells and induces apoptosis (17). According to this previous study, our observation implies that the deletion in ORF7a might affect the SARS-CoV-2 pathogenesis. However, further studies are needed to characterize the mechanisms underlying ORF7a functional alteration.
DISCUSSION
We successfully isolated SARS-CoV-2 from COVID-19 patients in the southwestern region of South Korea. According to viral clade classification based on GISAID data, SARS-CoV-2 in Gwangju mainly belong to the V and G clades. More specifically, during the initial breakout in February 2020, the V clade was major clade type, mainly due to spread in the Shincheonji religious group. Since July 2020, the GH clade, related to door-to-door sales, became the prevalent type. In addition, we identified 21 non-synonymous mutations and one frameshift deletion. This study provided genomic mutation analysis in SARS-CoV-2 isolated from patients in Gwangju, South Korea. It is important to analyze and share the genome characteristics of SARS-CoV-2 for interpreting alterations in infectivity, pathogenicity, and host-virus interactions by continuous monitoring of the genetic mutations. Future studies including additional genome sequences are needed to define the further evolution of the SARS-CoV-2 epidemic in Gwangju.
Lastly, as the rate of appearance of SARS-CoV-2 virus mutant is very fast and diversified in recent years, to avoid another SARS-CoV-2 pandemic, an ongoing effort is needed to monitor, collect and analyze data on new SARS-CoV-2 variants.
