

INTRODUCTION
Antibiotics are considered a milestone in the history of humanity and modern medicine as they are essential and life-saving weapons against numerous infectious diseases. Moreover, antibiotics are among the most effective chemotherapies in medical history (1). However, antibiotic resistance is becoming a major global health issue (2). Despite the popular notion that antibiotic exposure is limited to the modern “antibiotic era,” the researcher has discovered traces of exposed tetracycline in human skeletal remains dating back to 350–550 CE in ancient Sudanese Nubia (1, 3, 4). Nevertheless, in the modern era, after its discovery in the 1940s, tetracycline has taken a special place because of its antibacterial activity against many life-threatening microorganisms (5, 6).
Chlortetracycline was the first tetracycline discovered by the American Cyanamide Company in the 1940s and it is a natural tetracycline isolated from Streptomyces aureofaciens(4, 5, 6). Its effectiveness against various pathogens, such as Actinomycetes, Escherichia coli, Mycoplasma spp., and Rickettsiae has solidified its place as a key antibiotics in the treatment of infections (5). However, the increase in tetracycline resistance has necessitated the urgent development of a novel drug capable of counteracting this type of resistance (2). Following previous footprints, several generations of tetracycline drugs have been developed because of increased bacterial resistance, which is difficult to control. Pathogens such as Enterococci show vancomycin resistance, and Staphylococcus aureus shows methicillin resistance; thus, it has become difficult to control these multidrug-resistant microorganisms (7). Antibiotic resistance is a serious global crisis and a significant threat to public health (8, 9, 10). This unexpected resistance is too important for developing antibiotics to maintain public healthcare systems (8). The growing number of infectious agents produced by multidrug-resistant microorganisms poses a significant threat to the public healthcare system (11). Therefore, controlling multidrug-resistant (MDR) pathogens and keeping the public healthcare system safe force us to take significant action in developing antibacterial agents (8, 12). Moreover, developing new antibiotics through minor modifications or changes to existing classes of drugs is more feasible than creating an entirely new drug, which is complicated and time-consuming.
Eravacycline (TP-434) is a promising new fluorocycline antibacterial agent synthesized using a fully synthetic approach (13, 14). This methodology was initially introduced by Charest et al., showing its potential for the efficient production of this compound (15). Then, it was developed by Tetraphase Pharmaceuticals, Inc. (Watertown, MA, USA) as Xerava™ (15, 16) through a small alternation in the tetracyclic D-ring (17). An advanced generation of tetracycline-class drugs is synthesized by slight alterations at the C7 and C9 positions of the tetracycline D-ring, producing a new, fully synthetic fluorocycline. It was effective in phase 3 clinical trials, and the FDA approved it for intra-abdominal infections (cIAIs) with serious complications in 2018 (16. 17. 18). This has become a blessing for the healthcare system because cIAIs are the most common infections, causing significant morbidity and increasing healthcare expenses (12). The primary purpose of eravacycline is to surpass acquired resistance toward standard tetracycline class drugs linked to efflux pumping and a protective mechanism of bacterial ribosomes, which was found to surpass that resistance (18). Many countries, including the EU and USA, have permitted the intravenous administration of eravacycline for cIAI treatment owing to its efficacious action. However, it has not been approved for complicated urinary tract infection treatment because of poor outcomes and adverse events in clinical tests (18). In this review, we focus on the therapeutic application of eravacycline in the treatment of diseases caused by microorganisms that are resistant to conventional antibiotics.
CHEMISTRY AND STRUCTURAL CHARACTERISTICS
Eravacycline is a unique synthetic fluorocycline antibacterial agent designed to overcome resistance against typical tetracycline-specific efflux pumping and protective mechanisms of bacterial ribosomes (19, 20, 21). The molecular weight of eravacycline is 558.6 g/mol (2, 22). Structurally, eravacycline has an original tetracyclic D-ring structure containing oxygen atom substitutions at the C1, C3, C10, C11, C12, and C-12a positions, and dimethylamine groups at the C4, C5, and C6 positions, which are replaced with diverse substituents (Fig. 1A) (6). The attachment of fluorine atoms and pyrrolidinoacetamo groups at C7 and C9, respectively, produces 7-fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline (C27H31FN4O8), which is neither a naturally occurring nor semisynthetic tetracycline (6, 18, 22). Attachment of the fluorine atom (a weak polar group) at the C7 position increases the antimicrobial action through electron withdrawal to the aromatic ring. Moreover, substituting the pyrrolidinoacetamo group (a highly polar group with an essential nature) at position C9 enhances the surface area of eravacycline (21).

Fig. 1
Chemical structure of eravacycline (A) and tigecycline (B) showing alteration at C-7 and C-9 position.
This new fluorocycline structurally resembles tigecycline (TIG) (23). Tigecycline is a modified minocycline derivative used to combat bacterial tetracycline resistance mechanisms (24). Before eravacycline, TIG was the most effective tetracycline-class drug, with dimethylamino- and N-tert-butyl-glycylamido groups at the C7 and C9 positions, respectively (Fig. 1B) (25, 26). Eravacycline differs from TIG because of two changes in the D-ring structure upon substituting the pyrrolidine ring at the C9 position (23). This substitution enhances the antibacterial properties of eravacycline compared with TIG because the heteroaromatic ring is more sustainable than any cyclic alkylamine, reducing the resistance mechanism of the pathogen (21). This unique alteration stabilizes eravacycline more than other tetracycline-class drugs and increases its efficacy against gram-positive and gram-negative bacteria by overcoming bacterial-acquired resistance mechanisms against tetracyclines (21).
MODE OF ACTION
Tetracyclines exert their antibacterial action by attaching to the bacterial ribosome and inhibiting protein translation (27). Eravacycline also acts by incorporating the bacterial 30S subunit within the ribosome and inhibiting the connection of charged tRNA (aminoacyl tRNA) to the acceptor of the mRNA-ribosomal complex (18, 21). Thus, various linked amino acid residues are prevented from extending into polypeptide chains (18). Thus, bacterial protein synthesis is inhibited (21). However, this fluorocycline is different from previous cohorts of tetracycline-class drugs in that it is a synthetic antibiotic containing fluorine atoms (showing weak polarity) as well as a pyrrolidinoacetamo (showing high polarity with a slight essential nature) group on the ring D structure (5, 21). These minor alterations lead to a protective action against resistance mechanisms to specific tetracycline-class drugs in gram-positive and gram-negative bacterial species (28). All classes of tetracyclines, including fluorocycline, typically exhibit bacteriostatic activity. It also elicits slaughtering effects against certain strains of Acinetobacter baumannii (nosocomial infections), Klebsiella pneumoniae, and E. coli(6, 17).
PHARMACOKINETICS AND PHARMACODYNAMICS
Eravacycline was administered intravenously (i.v) with a weight-based strategy at 1 mg/kg twice daily with a definite infusion period of 60 min (17, 21, 29). The summary of the pharmacokinetic properties of eravacycline is presented in Table 1(6, 17). Formerly, orally formulated eravacycline was discontinued because of poor results in a phase 3 clinical survey of complicated urinary tract infections treatment (5). However, orally formulated dosage forms are still being developed (30). At the end of the parenteral dosage form, individual and repeated doses of IV eravacycline were administered at a dose of 1 mg/kg twice daily. The maximum plasma concentration (Cmax) value was 2125 ng/mL on the 1st day and 1825 ng/mL on the 10th day (2), with 12 h area under the curve (AUC) (0–12 h) values of 4305 ng h/mL and 6309 ng h/mL, respectively (5, 31). An increased plasma concentration increased the protein-binding portion from 79% to 90% (18). At steady state, eravacycline was significantly distributed, with a mean volume of distribution (Vd) of 321 L (5). Therefore, a high Vd value may be less effective for initial bloodstream infections (5). In human plasma and urine, unchanged eravacycline is a significant element of therapeutic products (32). It is mainly metabolized by the CYP3A4 enzyme and the flavin-containing mono-oxygenase-mediated oxidation pathway (5, 18). The primary metabolites of eravacycline are produced by the pyrrolidine ring (TP-6208) and pimerization at the C4 position, and another metabolite is TP-034 (5, 31). None of these metabolites show antimicrobial activity (32). Eravacycline is expelled from the body through urine (34%) and fecal waste (47%) as an intact drug, as well as metabolized products (18). Its elimination half-life was 20 h (5). A previous study showed that the AUC and Cmax increased slightly by 4% and 8.8%, respectively, in renally impaired patients; therefore, there is no need for renal dose adjustment (5, 17). Nevertheless, this drug exhibits extensively increased AUC (110.3%) and Cmax (19.7%) in severely hepatic-impaired patients (Child-Pugh class C); therefore, dose alteration is highly recommended with a limit of 1 mg/kg of weight twice daily intravenously on the first day of treatment, followed by 1 mg/kg once daily (5). Moreover, it has been shown to interact with a CYP3A4 inducer or inhibitor, which decreases its AUC value by 35%; thus, the dose of eravacycline should be increased to 1.5 mg/kg twice daily (18). Pharmacokinetic studies of orally administered eravacycline demonstrated 28% bioavailability and interaction with food elements, which decreased further absorption (33).
Table 1.
Summarized pharmacokinetic properties of eravacycline
Pharmacodynamics describes the relationship between antibacterial exposure and the microbiological response (6). Pharmacodynamic studies are essential to select an accurate dose of antimicrobials to treat infectious diseases (34). Its pharmacodynamic propeller efficiency is expressed as the ratio of the AUC of the unbound drug to the minimum inhibitory concentration (MIC) of the organism (AUC/MIC) (35, 36). In the following tables, it is shown that eravacycline exhibits wide-spectrum in vitro activity with MIC90 (MIC that inhibits 90% bacterial growth) of ≤ 2 mg/mL counter to the maximum pathogenic strains (6, 19). Table 2 shows eravacycline potency with an MIC90 value of ≤ 0.25 mg/L for all Staphylococci, Streptococci, and Enterococci(6, 17, 18, 19). Table 3 shows the antibacterial action of eravacycline against different types of aerobic and anaerobic gram-negative bacteria, including Enterobacteriaceae, Enterobacter cloacae, E. coli, K. pneumonia, Bacteroides ovatus, with MIC90 values not exceeding 2 mg/mL (18, 19, 37, 38). Moreover, Table 4 shows the in vitro action of eravacycline as well as its comparative drugs counter to the susceptibility of some resistant organisms, including Enterobacteriaceae, Citrobacter freundii MDR, K. pneumoniae MDR, and A. baumannii, which demonstrate eravacycline is two to eight-fold stronger than tigecycline against most of the pathogens (5, 6, 37, 39).
Table 2.
Eravacycline MIC90 values against some gram-positive bacteria
| Microbes | Number of total isolates (n) | MIC90 (μg/mL) | Reference |
|---|---|---|---|
| Aerobes | |||
| Enterococcus spp. | 406 | 0.06 | (18) |
| Enterococcus faecalis | 1605 | 0.06 - 0.12 | (17, 18) |
| E. faecalis (Vancomycin- resistant) | 108 | 0.12 | (18) |
| E. faecalis (Vancomycin-sensitive) | 121 | 0.12 | (18) |
| Staphylococcus aureus | 2024 | 0.12 | (17, 18) |
| Methicillin-resistant S. aureus (MRSA) | 2247 | 0.06 - 0.12 | (17) |
| Methicillin-susceptible S. aureus (MSSA) | 2006 | 0.06 - 0.25 | (17) |
| Streptococcus anginosus | 47 | 0.03 | (19) |
| Streptococcus mitis | 62 | 0.12 | (19) |
| Streptococcus intermedius | 31 | 0.06 | (19) |
| Anaerobes | |||
| Clostridium perfringens | 15 | 1 | (18) |
Table 3.
Eravacycline MIC90 values against some of gram-negative bacteria
| Microbes | Number of total isolates (n) | MIC90 (μg/mL) | Reference |
|---|---|---|---|
| Aerobes | |||
| Enterobacter spp. | 1268 | 0.5 | (37) |
| E. cloacae | 966 | 0.5-2 | (18) |
| E. cloacae MDR | 107 | 0.25 | (37) |
| Citrobacter freundii | 134 | 1-2 | (19, 38) |
| Enterobacteriaceae | 15,240 | 2 | (18) |
| Enterobacteriaceae CR | 623 | 2 | (18) |
| Enterobacteriaceae ESBL (+) | 179 | 1-2 | (18) |
| MDR enterobacteriaceae | 1235 | 2 | (18) |
| Escherichia coli | 4575 | 0.25-0.5 | (18) |
| E. coli MDR | 107 | 0.25 | (37) |
| E. coli ESBL (+) | 141 | 0.5 | (38) |
| E. coli ESBL (−) | 1036 | 0.25 | (38) |
| Klebsiella spp. | 1388 | 1 | (18) |
| Klebsiella oxytoca | 136 | 0.5-1 | (19, 38) |
| Klebsiella pneumoniae | 1853 | 1-2 | (18) |
| K. pneumoniae MDR | 138 | 2 | (37) |
| K. pneumoniae ESBL (+) | 21 | 2 | (38) |
| K. pneumoniae ESBL (−) | 360 | 0.5 | (38) |
| Anaerobes | |||
| Bacteroides caccae | 10 | 0.5 | (18, 37) |
| Bacteroides fragilis group | 286 | 1 | (37) |
| B. fragilis | 110 | 1 | (37) |
| B. ovatus | 30 | 1 | (37) |
| B. thetaiotaomicron | 70 | 1 | (37) |
| B. uniformis | 15 | 1 | (18, 37) |
| B. vulgatus | 18 | 0.5 | (37) |
| Parabacteroides distasonis | 26 | 1 | (37) |
Table 4.
Eravacycline and its comparators MIC ranges counter to different types of bacteria
| Drugs | Organism |
MIC50 value (mg/L) |
MIC90 value (mg/L) |
MIC ranges (mg/L) | Reference |
|---|---|---|---|---|---|
| Tigecycline | Enterobacteriaceae (n=3157) | 0.5 | 4 | 0.03 to 32 | (37) |
| Eravacycline | Enterobacteriaceae (n=3157) | 0.25 | 1 | 0.03 to 16 | (5) |
| Levofloxacin | Enterobacteriaceae (n=3157) | 0.06 | 8 | ≤0.004 to >8 | (37) |
| Ertapenem | Enterobacteriaceae (n=3157) | 0.015 | 0.25 | 0.004 to >2 | (37) |
| Eravacycline | MDR enterobacteriaceae (n=666) | 0.5 | 2 | 0.03 to 16 | (5) |
| Tigecycline | MDR enterobacteriaceae (n=666) | 1 | 4 | 0.06 to 32 | (37) |
| Meropenem | MDR enterobacteriaceae (n=666) | 0.06 | 0.5 | ≤0.004 to >4 | (37) |
| Eravacycline | Citrobacter freundii MDR (n=54) | 0.25 | 1 | max. 4 | (5, 37) |
| Tetracycline | Citrobacter freundii MDR (n=54) | 4 | >64 | >64 | (37) |
| Tigecycline | Citrobacter freundii MDR (n=54) | 0.5 | 2 | max. 4 | (37) |
| Minocycline | Citrobacter freundii MDR (n=54) | 4 | >16 | >16 | (37) |
| Eravacycline | E. cloacae MDR (n=95) | 0.5 | 2 | 0.12 to 4 | (5, 37) |
| Tigecycline | E. cloacae MDR (n=95) | 1 | 4 | max. 4 | (6, 37) |
| Eravacycline | E. coli MDR (n=15) | 0.12 | 0.25 | 0.03 to 2 | (5, 37) |
| Tigecycline | E. coli MDR (n=15) | 0.25 | 1 | max. 4 | (37) |
| Eravacycline | K. pneumoniae MDR (n=142) | 0.5 | 2 | 16 | (37) |
| Minocycline | K. pneumoniae MDR (n=142) | 8 | >16 | >16 | (37) |
| Tigecycline | K. pneumoniae MDR (n=142) | 1 | 4 | max. 8 | (6, 37) |
| Eravacycline | MSSA (n=256) | 0.06 | 0.12 | 0.03 to 0.5 | (37) |
| Tigecycline | MSSA (n=256) | 0.12 | 0.25 | 0.06 to 1 | (37) |
| Tetracycline | MRSA (n=256) | 0.25 | >16 | ≤0.06 to >16 | (37) |
| Eravacycline | MRSA (n=256) | 0.06 | 0.12 | 0.015 to 1 | (5) |
| Tigecycline | MRSA (n=256) | 0.25 | 0.5 | 0.06 to 2 | (37) |
| Eravacycline | E. faeciun and E. faecalis combined (n=397) | 0.06 | 0.06 | 0.008 to 0.5 | (5) |
| Tigecycline | E. faeciun and E. faecalis combined (n=397) | 0.12 | 1 | 0.03 to 2 | (37) |
| Eravacycline | S. maltophilia (n=469) | 1 | 2 | 0.06 to 8 | (5, 37) |
| Minocycline | S. maltophilia (n=469) | 1 | 2 | 0.25 to 64 | (37) |
| Tigecycline | S. maltophilia (n=469) | 2 | 4 | 0.25 to >16 | (37) |
| Eravacycline | A. baumannii (n=503) | 0.5 | 1 | ≤ 0.015 to 16 | (37) |
| Minocycline | A. baumannii (n=503) | 2 | 16 | 0.06 to >64 | (37) |
| Tigecycline | A. baumannii (n=503) | 4 | 8 | 0.12 to >16 | (37) |
| Eravacycline | H. influenzae | 0.12 | 0.25 | ≤0.015 to 0.5 | (6) |
| Delafloxacin | H. influenzae | 0.001 | 0.004 | ≤0.001 to 0.25 | (39) |
| Levofloxacin | H. influenzae | 0.015 | 0.03 | 0.008 to > 2 | (39) |
DOSES AND ADMINISTRATION
Eravacycline, administered intravenously, is approved in the EU (32) and USA for cIAIs treatment in patients over 18 years of age (17). This is a weight-based dosing system that requires no drug monitoring (40). For intravenously administered eravacycline, 1 mg/kg was allowed for 4–14 days (5, 17, 32). However, the period of therapy may be changed by the intensity and site of infection in the body and depends on the clinical feedback of the patients (40).
TOLERABILITY PROFILE AND ADVERSE EFFECTS
After several research trials, a suitable tolerability profile was found in intravenously administered eravacycline patients with cIAIs who participated in phase 2 and 3 trials, with most treatment-involving adverse events (41, 42). Initially, safety and tolerability tests were assessed in phase 1 orally given solo dosing study with six healthy volunteers in three sets taking 0.2 g or 0.3 g or placebo, respectively (6). Reactions at the infusion site (7.7%), nausea (6.5%), and vomiting (3.7%) were the most common adverse reactions experienced in the trials (17). Adverse effects such as reactions at the infusion site comprising discomfort or aching; erythema, swelling, or inflammation; and phlebitis or superficial thrombophlebitis have also been reported in phase 2 and 3 trials (17, 32). Approximately 2% of the patients (n = 520) discontinued therapy due to gastrointestinal disorders (18). Nevertheless, no patient discontinued the therapy for infusion site reactions because it could be reduced by decreasing the infused concentration, otherwise diminishing the infusion speed (18, 32). Eravacycline may exhibit adverse reactions such as photosensitivity, anti-anabolic activity, and pseudotumour cerebi, leading to augmented blood urea nitrogen levels, acidosis, azotemia, pancreatitis, and abnormal liver function assessments (17, 32). Severe hypersensitivity reactions can be induced with eravacycline; therefore, it is forbidden to use this drug in patients who experienced hypersensitive reactions before the tetracycline class of drugs (18). During pregnancy, consumption of eravacycline during tooth development in the fetus may cause permanent tooth discoloration and enamel hypoplasia (17, 32). Bone growth can be reversibly inhibited (18). Some adverse events are discussed below using a simple pie chart (Fig. 2).
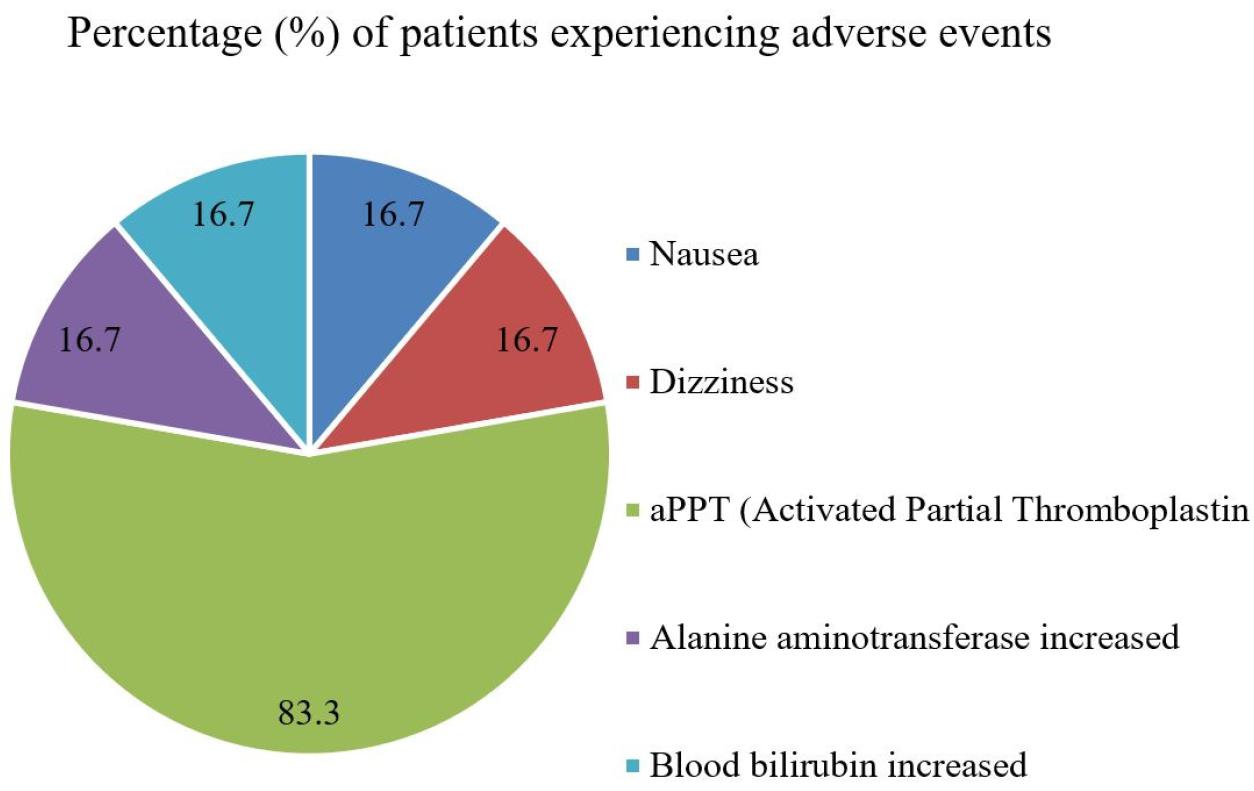
Fig. 2
Percentage (%) of patients experiencing adverse events when patients taken eravacycline 0.2 g and 0.3 g daily. 83.3% of patients experienced activated partial thromboplastin (aPPT) and 16.7% of patients experienced nausea, dizziness, increased alanine aminotransferase and blood bilirubin, respectively.
RESISTANCE AND DRUG INTERACTION
Eravacycline demonstrated in vitro action against carbapenem-resistant Enterobacteriaceae through the appearance of specific β-lactamases and AmpC β-lactamase enzymes (5, 18). Eravacycline is also effective against most gram-negative bacteria (21). It overcomes the resistance mechanism of bacteria by inhibiting bacterial protein synthesis and maintaining the efflux pumping of drugs outside the bacterial strains (6, 30, 43). It also acts against a few β-lactamases and acts on organisms through other mechanisms (31). Moreover, it kills some bacterial strains linked to upregulated and nonspecific intrinsic MDR efflux pumping mechanisms and targets spot alterations in 16S ribosomal RNA or specific ribosomal 30S proteins (5, 18). Eravacycline kills Enterococcus spp. that harbor mutations encoded by the microbial rpsJ gene (18, 32). However, eravacycline does not show target-oriented cross-activity with other antibiotics, such as fluoroquinolones, penicillins, carbapenems, or cephalosporins (44). Grossman et al. indicated that efflux pumps, mainly tetA, tetB, tetK, and ribosomal protective protein genes, such as tetM and tetQ, are common tetracycline resistance genes that usually do not affect the efficacy of eravacycline (45). However, it may have minimal effects via other mechanisms identified in future studies (5, 6, 18). Another concern is that K. pneumoniae shows resistance to eravacycline through overexpression of efflux pumps, such as MacAB and OqxAB, and by the transcriptional regulatory gene (ramA), which increases the MIC value of eravacycline in some investigated K. pneumoniae isolates treated with eravacycline (44).
Several drug-drug interaction reports have investigated the absorption, distribution, metabolism, and excretion (ADME) profile of intravenously formulated eravacycline with other drugs (31). Eravacycline elicits activity with potent CYP3A4 inducers, such as carbamazepine, rifampin, and phenytoin (5, 18). This results in an increased metabolic rate and extent of eravacycline activity (20, 31). It also interacts with the activity of CYP3A4 inhibitors such as itraconazole and clarithromycin, which decreases its efficacy (31). To address this issue, the eravacycline dose should be increased by 50% when administering potent CYP3A4 inducers or inhibitors by exceeding 1.5 mg/kg from 1 mg/kg twice daily (5, 18). Eravacycline also inhibits the metabolism of anticoagulants (such as warfarin) and plasma prothrombin activity (5, 46). In addition to the efficacy of orally administered retinoid medicines (such as isotretinoin and acitretin), amoxicillin and ampicillin activities may be reduced when eravacycline is administered (40, 41).
CONCLUSION
Eravacycline opens new accessibility to clinicians as a non-β lactam choice counter to various gram-negative organisms showing β-lactam resistance (5). It also shows intense activity against a broad spectrum of clinically related gram-positive and gram-negative aerobic and anaerobic bacterial species (45, 47), comprising carbapenem-resistant Enterobacteriaceae, E. coli, K. pneumonia, B. ovatus, and organisms that show prior classes of tetracycline, vancomycin, and methicillin resistance by specific resistance mechanisms (18, 48). Currently, eravacycline holds a new therapeutic position as an antibacterial agent in Clostridium difficile-infected patients, but its main focus is cIAIs treatment (aged over 18 years) (49). It shows more potent in vitro efficacy with an excellent tolerability index than other tetracyclines (e.g., tigecycline), making it more innovative, especially when treatment with resistant pathogens is necessary (46). Finally, eravacycline is a promising broad-spectrum fluorocycline that shows the best efficacy when administered intravenously (50). Various infectious diseases caused by bacterial antibiotic resistance are increasing at a high rate globally (14). Therefore, this novel fluorocycline should be appropriately guided to defend against serious resistance mechanisms, similar to the previous empirical options.
