INTRODUCTION
Helicobacter pylori (H. pylori) is a gram-negative, spiral-shaped bacterium that grows under microaerobic or capnophilic environment (1, 2, 3, 4, 5). It colonizes the gastric mucosa by adhering to the epithelial surface and exhibits remarkable adaptability that enables long-term persistence (6, 7). The strong urease activity and motility of H. pylori is advantageous to survive in the gastric mucosa (8, 9, 10, 11, 12, 13). In particular, urease-mediated neutralization of gastric acid by H. pylori induces hyperacidity and mucosal injury (14, 15), leading to persistent inflammation, which in turn can cause acute or chronic gastritis, peptic ulcers, and gastric cancers in humans (16, 17, 18, 19).
Dendritic cells (DCs) are not only responsible for initiating adaptive immune responses but also serve as key antigen-presenting cells that determine the overall direction of the immune response. Upon recognition of H. pylori, naïve DCs internalize the bacterium and undergo maturation, leading to antigen presentation and T-cell activation (20). Responding to the immune reaction of DCs, H. pylori modulates the immune response of DCs to evade the immune clearance through various mechanisms, thereby establishing chronic infection (20, 21). Indeed, mature DCs secrete IL-1β and IL-6, which promote the differentiation of Th17 cells (21, 22, 23). These DCs may also induce Th1 immune responses characterized by the production of IFN-γ, MHC II molecules, and IL-12α (22, 24). On the other hand, H. pylori skewed the immune responses of DCs to induce the expression of TGF-β and the transcription factor FoxP3, thereby facilitating the differentiation of regulatory T cells (Tregs), which produce IL-10 and suppress Th1-mediated inflammation. Thus, DC-mediated antigen presentation in H. pylori infection can lead to divergent immunological outcomes, ranging from inflammatory clearance via Th17/Th1 pathways to immune tolerance mediated by Tregs (25, 26, 27).
Most clinical isolates of H. pylori grow poorly in vitro and are difficult to use in in vivo models such as mice or Mongolian gerbils, as the bacteria is typically adapted to human gastric mucosa (28, 29). Infection susceptibility varies by mouse strain and H. pylori genotype (30). Although the mouse-adapted Sydney Strain 1 (SS1) is widely used (31, 32), its genome has not been fully annotated, limiting its utility for genomic or epidemiological studies. Previous work identified two clinical isolates—H. pylori 51 strain (Hp51) and H. pylori (Hp52)—that successfully infect experimental mice (33). Proteomic analyses revealed increased expression of urease and catalase in Hp52, and upregulation of ATP-dependent protease binding subunits in Hp51, suggesting that differential protein expression contributes to varying infectivity across isolates. Hp51 (Accession CP000012.1) and Hp52 (Accession CP001680.1), isolated in Korea from gastric cancer and peptic ulcer patients, were fully sequenced and deposited in GenBank. In contrast, H. pylori strains 26695 (Hp26695) and J99 (HpJ99), despite having published genome sequences (34, 35), showed very low level of infectivity in mice and are therefore used primarily for in vitro studies.
To understand strain-specific infectivity, we asked two central questions: (i) why do H. pylori strains differ in their ability to infect mice, and (ii) what bacterial factors determine infectivity? To answer this question, it is necessary to perform a comparative analysis of gene expression among H. pylori strains with differing levels of infectivity, in order to construct a gene library of candidates suspected to contribute to the differences in infectivity. Additionally, the functional roles of each gene in the immune response to H. pylori must be elucidated.
A major barrier to answering these questions is the technical difficulty of genetically manipulating H. pylori. Knockout (K/O) strains are generated by inserting antibiotic resistance markers such as chloramphenicol resistance (cat) or kanamycin resistance (aphA-3) into open reading frames (ORFs) via cross-over PCR (36). However, compared with E. coli, H. pylori requires stringent growth conditions, and standard natural transformation or electroporation methods are often ineffective (37, 38). To overcome these barriers, improved transformation and culturing methods need to be employed. H. pylori transformation efficiency was improved by modifying growth supplements and optimizing the timing of DNA exposure under established culture conditions (38, 39). Growth of H. pylori typically requires supplementation with bovine or horse serum (40, 41, 42), and environmental factors such as cell division rate, temperature, supply level of O2 or CO2, and nutrient availability strongly influence bacterial adaptation and colonization (43). Essential growth factors—including dextrose, sodium chloride, and glucose—support survival in the gastric environment (44, 45, 46, 47). As H. pylori divides more slowly than E. coli or the resident gastric microbiota (48), exogenous nutrient supplementation such as Albumin–Dextrose–Saline (ADS) is required, similar to Mycobacterium spp. (49, 50). Therefore, ADS was used as a serum substitute to enhance the transformation efficiency of H. pylori.
In this study, we compared the gene expression profiles of H. pylori strains Hp52, Hp26695, and HpJ99—which exhibit distinct levels of infectivity in mice—using microarray analysis, and constructed a gene library comprising candidates presumed to contribute to these differences in infectivity. To investigate the immunological roles of these genes during H. pylori–host interactions, we generated individual gene-K/O mutants and analyzed DC immune responses to these mutants in mice. Through this approach, we aimed to identify genes associated with the infectivity-related immune modulation in H. pylori.
MATERIALS AND METHODS
Culture of H. pylori
All H. pylori strains were provided by Fastidious Specialized Pathogen Resources Bank of Gyeongsang National University Hospital (Korea). Hp52 was isolated from a gastric cancer patient and possesses infectivity in mice. In contrast, the standard strains used in the experiment, Hp26695 and HpJ99, do not possess infectivity in mice. Half-thawed H. pylori stocks were inoculated onto Brucella agar plates (MP Biomedicals, Korea) supplemented with 10% horse serum (Gibco, USA) and incubated at 37°C under a microaerophilic atmosphere containing 10% CO₂ for 48 h. Following primary isolation, the bacteria were subcultured in the growth medium for at least three consecutive passages at 24 h intervals. The cultured medium was reinoculated into 3 mL of fresh medium and incubated for 16 h using the thin-layer liquid culture method (39).
Microarray analysis using H. pylori total RNA
Total RNA was extracted from cultured H. pylori using the TRIzolTM total RNA extraction kit (ThermoFisher, USA), and microarray analysis was performed (Genomictree Inc., Korea). Total RNA was quantified and amplified using Agilent’s Low RNA Input Linear Amplification Kit PLUS (Agilent, USA), and subsequently labeled by the addition of 0.5 μL of Cyanine 3-CTP (Cy3) according to the manufacturer’s instructions. Hybridization was carried out for 17 h at 65°C using Agilent’s Gene Expression Hybridization Kit (Agilent, USA). The Cy3-hybridized samples were scanned using the SureScan Microarray Scanner (Agilent, USA), and fluorescence signals were extracted using Agilent’s Feature Extraction Software. In this study, Hp52 was used as the reference strain. For each gene, probe recognition was achieved by aligning to a 120 bp region beginning at the N-terminus of the mRNA. Whole-genome clustering of H. pylori genes was performed using Agilent’s GeneSpring Software (Agilent, USA). Differential gene expression between Hp52, Hp26695, and HpJ99 was assessed, and genes exhibiting statistically significant changes were identified using an ANOVA t-test.
Verification of microarray results
All genes that showed significantly different expression in Hp26695 and HpJ99 compared to the Hp52 in the microarray analysis were verified using quantitative real-time RT-PCR (qRT-PCR) . The nucleotide sequences of selected target genes, along with the housekeeping gene (16S rRNA), were retrieved from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). Primer sequences were designed using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and PrimerSelect® (DNASTAR, USA). Total RNA was extracted from cultured H. pylori using the TRIzol™ Reagent (Invitrogen, USA). Complementary DNA (cDNA) was synthesized using the GoScript™ Reverse Transcription System (Promega, USA) with the following thermal cycling conditions: 5 min at 25°C, 1 h at 42°C, and 15 min at 70°C. qRT-PCR was performed with 2× SsoFast EvaGreen Supermix (Bio-Rad, USA) and 10 pmol/μL of primers specific to either the 16S rRNA housekeeping gene or the target gene using Rota-Gene Q (Qiagen, USA).
Construction of Gene K/O Mutants
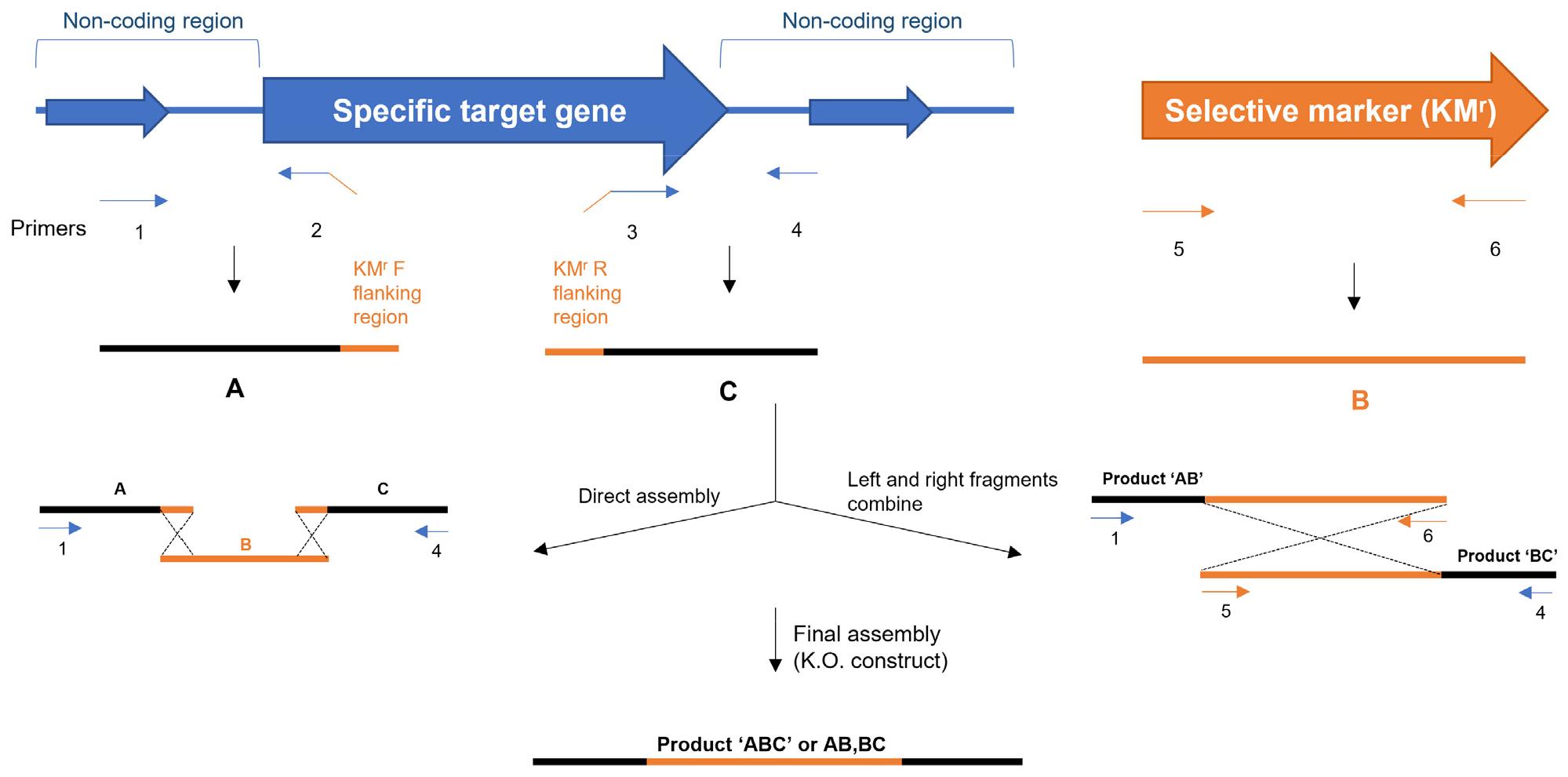
Genes that showed differential expression in the microarray analysis and qRT-PCR validation were further examined using NCBI’s BLAST (Basic Local Alignment Search Tool). For each selected gene within the Hp52 genome registered in NCBI, the corresponding nucleotide regions were edited using sequence-manipulation software, including Lasergene (DNASTAR, USA) and SnapGene (GSL Biotech LLC, USA). Approximately 20 bp at the N-terminal region of each target gene were defined as the upstream flanking sequence, whereas 20–25 bp at the C-terminal region were designated as the downstream recognition sequence. All designed sequences were adjusted to achieve a G/C content of 35–50%. The N-terminal non-coding region of each gene, spanning 30–70 bp from the start codon, was selected to generate gene-specific mutant fragments of approximately 200 bp. A kanamycin-resistance (KMr) marker was inserted into the open reading frame (ORF) of each gene. The forward primer for the KMr cassette was positioned downstream of the upstream flanking sequence, whereas the reverse primer for the KMr cassette was positioned upstream of the downstream recognition sequence. K/O constructs were generated by sequential PCR using a cross-over assembly strategy. The flanking regions and the KMr cassette were assembled using two alternative approaches. In the direct-assembly method, the upstream and downstream flanking sequences were combined with the mutant fragment and the KMr cassette in a single PCR reaction to produce the complete K/O construct. Alternatively, the components were amplified as separate segments: a left-arm fragment consisting of the upstream flanking sequence fused to the KMr cassette, and a right-arm fragment consisting of the KMr cassette fused to the downstream flanking sequence. These fragments were then joined during a final assembly step. The resulting K/O constructs, generated either by direct assembly or by combining the left- and right-arm segments, were subsequently used for natural transformation of H. pylori.
PCR Conditions for Amplification and Assembly of K/O Constructs
PCR amplification of the upstream and downstream flanking fragments was carried out using an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 7 min. These flanking fragments were generated using a Taq polymerase pre-mix kit (BIONEER, Korea). Amplification of the KMr cassette was performed using a Pfu polymerase kit (ELPIS Biotech, Korea) with an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 90 s, and a final extension at 72°C for 7 min. All mutant fragments and KMr cassettes were assembled using a cross-over PCR method, as illustrated in Supplementary Fig. 1. Final amplification of the K/O constructs was performed using Taq DNA polymerase with an initial denaturation at 94°C for 5 min, followed by 25 cycles of 94°C for 40 s, 60°C for 40 s, and 72°C for 2 min. The sequences of all primers used in this study are listed in Table 1.
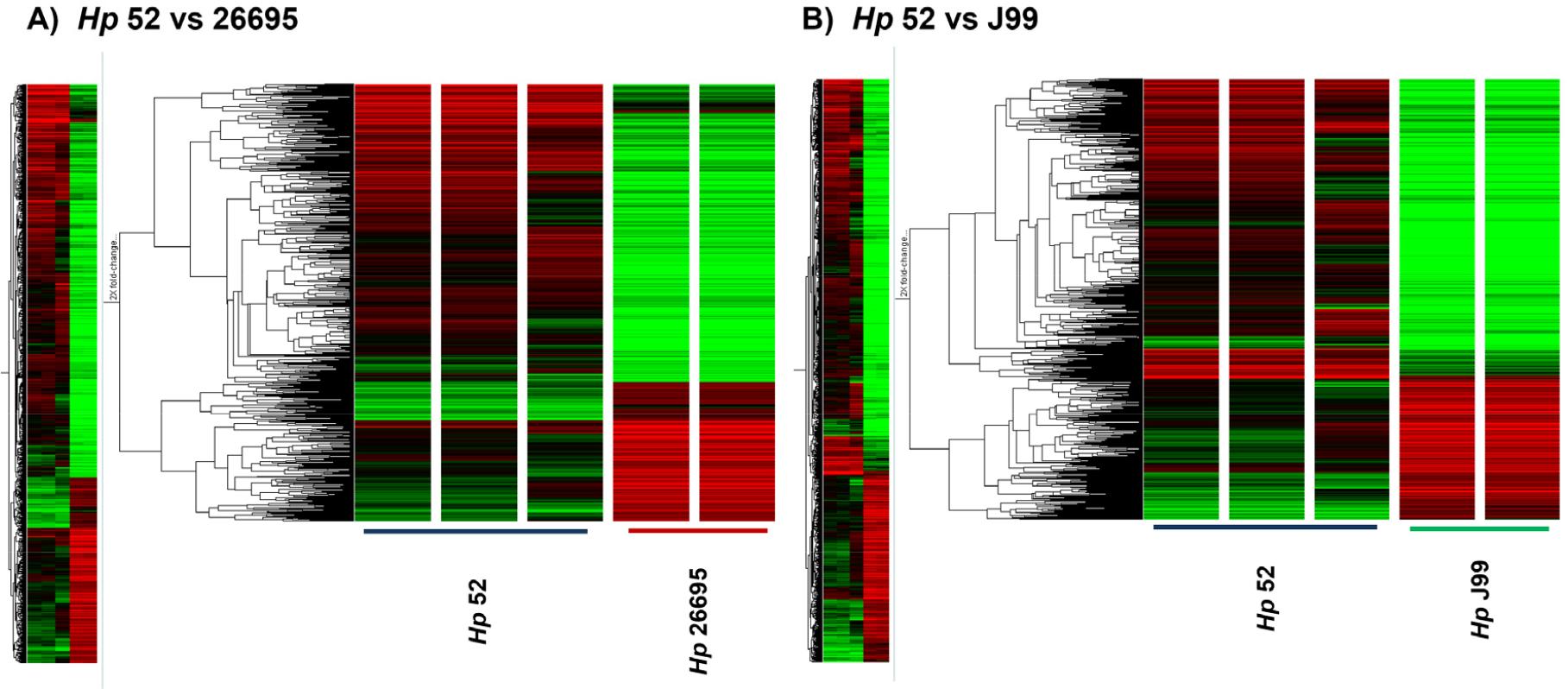
Fig. 1
Whole genome cluster of H. pylori using microarray analysis. A, Comparison of up- or down-regulation in gene expression between H. pylori 52 and 26695; B, Clustering of H. pylori 52 and HpJ99. The statistical significance of expression was determined by ANOVA t-test.
Table 1.
Primers information for amplifying knockout construct
Targeted Gene K/O via Natural Transformation
Natural transformation of H. pylori was performed using Brucella agar supplemented with 10% ADS enrichment (composed of 5% bovine serum albumin fraction V, 2% dextrose, and 0.85% sodium chloride). Hp52 cultured in liquid medium for 24 h was harvested and adjusted to a final absorbance of OD₆₀₀ = 1.0 (3.8 × 10⁸ CFU/mL). A 20µL aliquot of the bacterial suspension was spotted, without smearing, onto Brucella agar with ADS enrichment. The agar plates were pre-incubated at 37°C under 10% CO₂ for 12 h to allow bacterial adaptation. Linearized K/O constructs (1 µg in 10 µL) were then applied directly onto the pre-incubated H. pylori spots. After which, the bacterial cells were gently smeared within an approximate 10-mm diameter area and incubated under standard microaerophilic conditions to permit natural transformation.
Bone marrow–derived dendritic cells and infection assay for H. pylori mutants
Bone marrow–derived dendritic cells (BMDCs) were isolated from 6–7-week-old C57BL/6 mice (Koatech, Korea). Bone marrow cells were suspended in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS; MP Biomedicals, USA) and 1% penicillin–streptomycin (Gibco, USA). BMDC differentiation was induced by culturing the cells in RPMI 1640 containing 10% FBS, 10 ng/mL GM-CSF (PeproTech, Korea), and 5 ng/mL IL-4 (PeproTech, Korea). The cell suspension was seeded into T75 flasks (Corning, USA) at 10 mL per flask and incubated for 5 days at 37°C in a 5% CO₂ atmosphere, with medium changes performed every 2 days. Fresh GM-CSF and IL-4 were added at identical concentrations during each medium replacement. Differentiated BMDCs were adjusted to a density of 1 × 10⁶ cells/mL in T75 flasks with 10 mL of medium and were allowed to rest for 4 h at 37°C with 5% CO₂. Hp52 and mutant strains derived from Hp52 were added at a multiplicity of infection (MOI) of 10 and incubated with BMDCs for 6 h under the same conditions. GAPDH was used as the endogenous control gene. Expression levels of 11 cytokine- and activation-related genes—including TGF-β, IFN-γ, CCR7, MHC class II, IL-1β, IL-6, IL-10, IL-12α, CD40, CD80, and CD86—were quantified in Hp52–pulsed and mutant-pulsed BMDCs using Rota-Gene Q (Qiagen). Ct values were obtained using Rotor-Gene Q Series Software (Qiagen), and relative expression levels were compared across experimental groups by the 2-ΔΔCT method using the house-keeping gene, GAPDH. All animal experiments were conducted in accordance with the guidelines of the Gyeongsang National University Institutional Animal Care and Use Committee (IACUC) and were approved by the committee (protocol number: GNU-210901-M0074-01).
RESULTS
Differential gene expression profiles among Hp52, Hp26695, and HpJ99
All microarray analyses were performed using H. pylori RNA samples that met quality thresholds of OD₂₆₀/₂₈₀ ≥ 1.8 and OD₂₆₀/₂₃₀ ≥ 1.6. A total of 1,432 genes were detected across strains Hp52, Hp26695, and HpJ99 (Fig. 1). Comparative transcriptomic analysis between Hp52 and Hp26695 identified 21 genes that exhibited ≥2-fold expression changes across all probes. In the comparison between Hp52 and HpJ99, 41 genes demonstrated ≥2-fold differential expression. Among the significantly altered genes, 5 genes exhibited >5-fold down-regulation in Hp26695 compared to the Hp52. In strain HpJ99, 15 genes were down-regulated by >5-fold, excluding non-coding regions, compared to the Hp52 (Table 2). Notably, 2 genes—HPKB0424 (hypothetical protein) and HPKB1443 (ald)—showed commonly reduced expression in both Hp26695 and HpJ99. HPKB0424 and ald were down-regulated by 5.23-fold and 11.59-fold, respectively, in Hp26695, and by 24.12-fold and 22.77-fold, respectively, in HpJ99. In addition, 4 genes were identified that were not expressed in the HpJ99, including HPKB0346 (Hypothetical protein), HPKB0407 (Acetyl-CoA synthetase), HPKB0914 (Hypothetical protein), and HPKB1145 (Hypothetical protein) (Table 2).
Table 2.
Expression information of H. pylori classified through microarray analysis
Confirmation of expression reproducibility using real-time qRT-PCR
Validation of microarray results was performed based on the 16S rRNA–normalized expression levels using qRT-PCR. When compared with Hp52, consistent differential expression patterns were observed in Hp26695 and HpJ99. The expression of HPKB0424 was reduced by 4.03-fold in Hp26695 and by 7.83-fold in HpJ99 relative to Hp52 (p < 0.001). HPKB1443 (ald, encoding alanine dehydrogenase) also showed significantly decreased expression, with 2.45-fold and 3.04-fold reductions in Hp26695 and HpJ99, respectively (p < 0.001). Among the genes not expressed in HpJ99, HPKB0407 exhibited a 0.96-fold decrease in Hp26695, although the change was not statistically significant. In contrast, HPKB0346 showed a non-significant 0.70-fold increase in Hp26695. Based on these findings, four genes—HPKB0346, HPKB0407, HPKB0424, and ald—were identified as candidates displaying reproducible expression patterns across both microarray and RT-qPCR analyses or lacking detectable expression in HpJ99 (Fig. 2).
Production of recombinant knock-out constructs
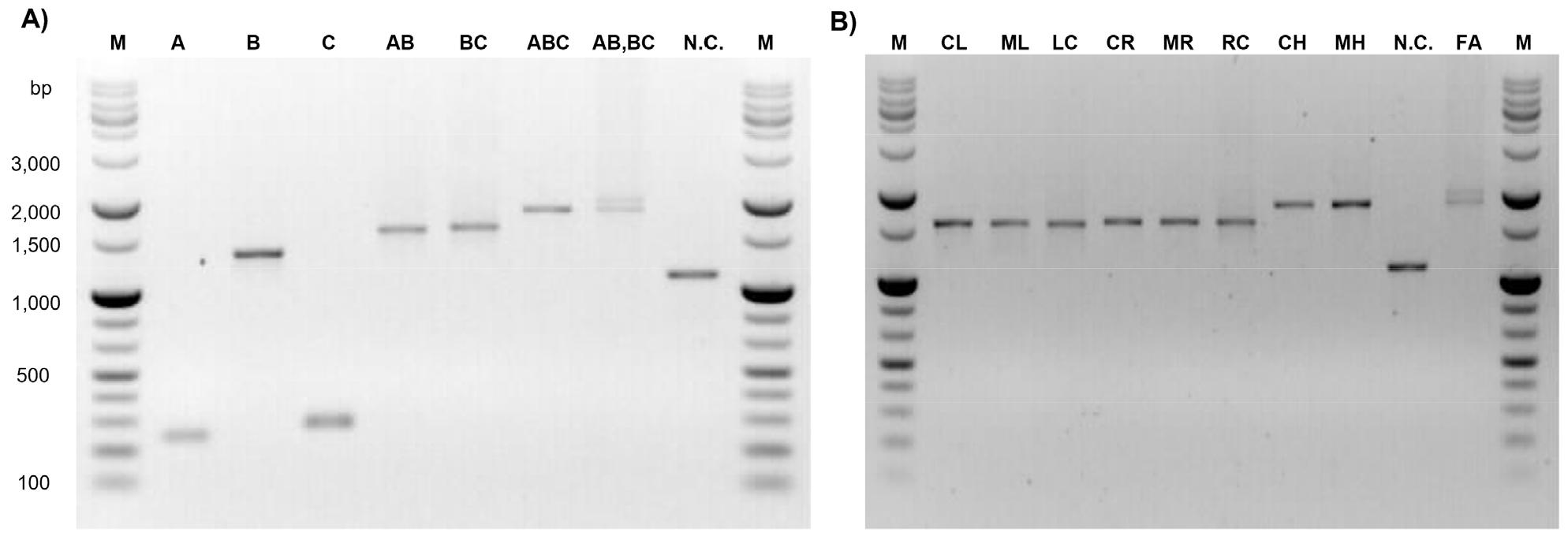
The K/O constructs incorporated the KMr cassette aphA-3 (1,401 bp). Four final assemblies were generated: ΔHPKB0346, ΔHPKB0407, ΔHPKB0424, and ΔHPKB1443 (Δald). The ΔHPKB0346 construct consisted of a 251 bp upstream fragment and a 234 bp downstream fragment, yielding a total assembly size of 1,751 bp. The ΔHPKB0407 construct was composed of a 230 bp upstream fragment and a 126 bp downstream fragment, with a final size of 1,622 bp. The ΔHPKB0424 construct included a 255 bp upstream fragment and a 392 bp downstream fragment, resulting in a total size of 2,004 bp. The Δald construct contained a 153 bp upstream fragment and a 341 bp downstream fragment, with a total size of 1,851 bp. Successful amplification and assembly of each K/O construct were verified by agarose gel electrophoresis (Supplementary Fig. 2).
Comparison of cytokine expression levels using 2-ΔΔCt analysis
Cytokine gene expressions in Hp52–pulsed DCs and K/O mutant–pulsed DCs were quantified using the 2−ΔΔCt method (Fig. 3). Relative to DCs pulsed with wild-type Hp52, IFN-γ expression was significantly reduced in all mutant groups, with fold-decreases of 1.47 (ΔHPKB0346), 1.76 (ΔHPKB0407), 1.89 (ΔHPKB0424), and 1.48 (Δald). MHC class II expression was also decreased in DCs pulsed with ΔHPKB0407 (2.36-fold), ΔHPKB0424 (2.72-fold), and Δald (2.23-fold) compared to the Hp52–pulsed DCs. Expression of IL-1β, IL-6, and IL-10 was down-regulated in DCs stimulated with ΔHPKB0346 (IL-1β: 2.08-fold; IL-6: 1.84-fold; IL-10: 4.44-fold), ΔHPKB0407 (IL-1β: 2.13-fold; IL-6: 1.42-fold; IL-10: 4.47-fold), and ΔHPKB0424 (IL-1β: 2.02-fold; IL-6: 2.22-fold; IL-10: 5.54-fold). CD40 expression was reduced in DCs pulsed with ΔHPKB0346 (1.69-fold), ΔHPKB0407 (1.74-fold), and ΔHPKB0424 (1.92-fold), while CD80 expression was decreased in DCs stimulated with ΔHPKB0346 (1.18-fold), ΔHPKB0407 (1.41-fold), and Δald (0.78-fold). In contrast, DCs pulsed with the Δald mutant exhibited marked increases in IL-1β (1.94-fold), IL-6 (2.62-fold), and IL-10 (2.38-fold) compared to the wild-type–pulsed DCs.
DISCUSSION
H. pylori possess numerous virulence determinants that contribute to its ability to establish colonization and persist within the gastrointestinal tract. Several factors have been implicated in infection establishment and inflammatory responses, including the well-characterized cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA), both of which are strongly associated with bacterial colonization and the risk of gastric cancer development (51, 52, 53, 54, 55). Additional virulence factors such as alpA, babA2, and hopZ have also been reported(56, 57, 58). These factors are thought to enhance colonization efficiency and promote infection-associated pathology (58); however, many factors that influence the establishment of infection have yet to be identified.
In this study, we aimed to identify virulence-associated genes involved in the establishment of H. pylori infection and the corresponding host immune responses. A comparative microarray analysis was conducted to examine global gene expression differences among three H. pylori strains—Hp52, which exhibits high infectivity in mice, and the less infectious strains Hp26695 and HpJ99. Four genes showing pronounced differential expression or presence–absence variation were identified. To investigate the contribution of these genes to colonization establishment, K/O mutants were generated in the Hp52 background. By assessing the immune responses of mouse BMDCs to each mutant, we inferred the functional roles of the selected genes in the establishment of H. pylori colonization.
Relative to strains Hp26695 and HpJ99, the Hp52 strain exhibited elevated expression or unique possession of four genes: HPKB1443 (ald), HPKB0346, HPKB0407, and HPKB0424. Among these, HPKB0407 encodes acetyl-CoA synthetase (ACS), which was absent in HpJ99. ACS is a key enzyme involved in acetate metabolism and TCA cycle function (59, 60). Although acetyl-CoA carboxylase (ACC) has been suggested to influence H. pylori growth under CO₂-enriched conditions (61), the role of ACS in H. pylori pathogenesis has not been previously demonstrated. Given their metabolic similarities, ACS may perform functions analogous to ACC, providing a potential explanation for its contribution to bacterial growth and infectivity. HPKB1443 encodes alanine dehydrogenase (Ald), an oxidoreductase that catalyzes the reversible deamination of L-alanine using NAD⁺ or NADP⁺ (62). In Bacillus subtilis, Ald is required for spore formation (63), and in Mycobacterium spp., it is secreted extracellularly and supports chronic persistence under nutrient-limited conditions (64, 65), making it a potential therapeutic target (66). These findings collectively suggest that ald may contribute to the establishment of chronic H. pylori infection.
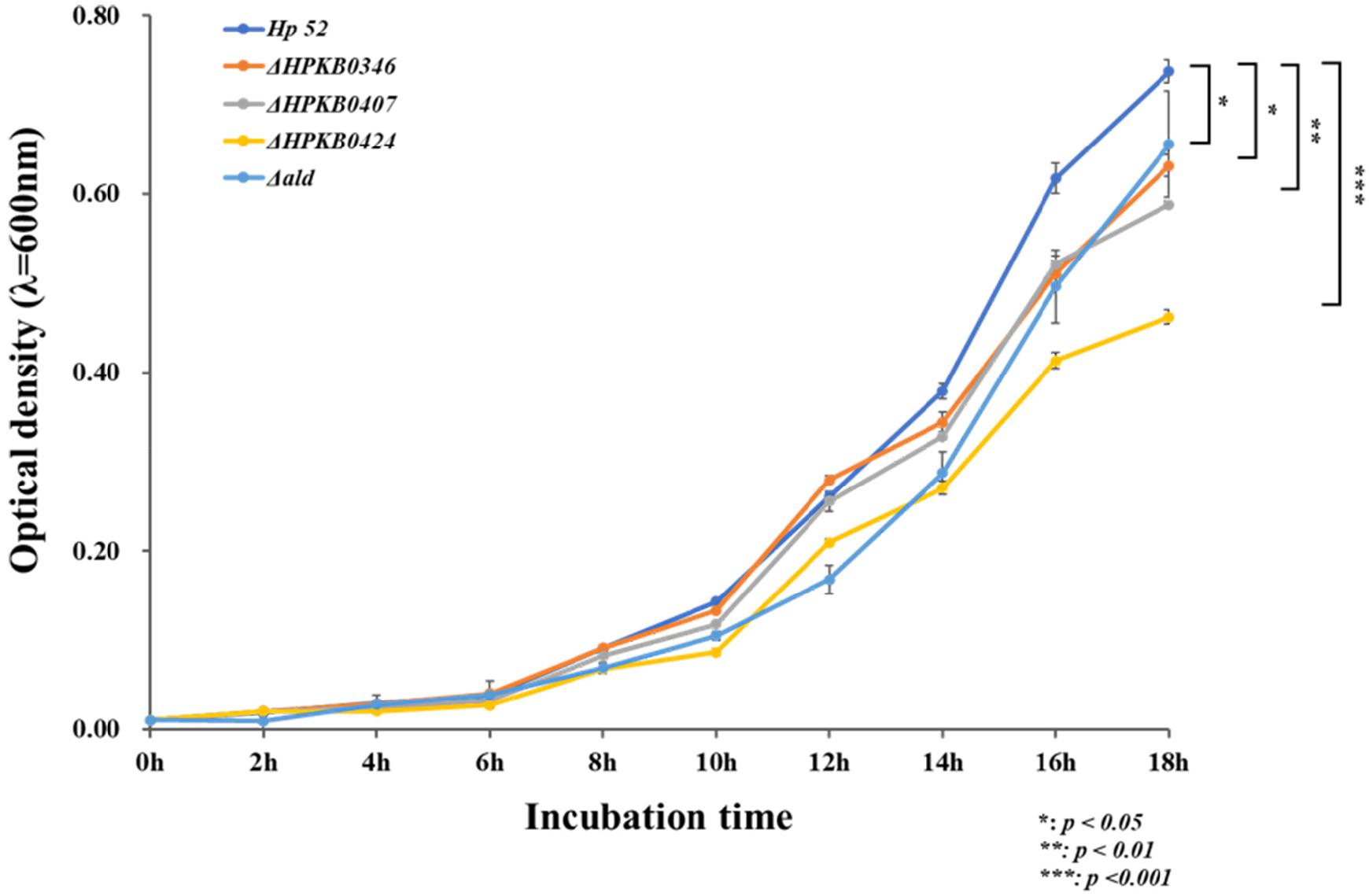
Although the functions of HPKB0346 and HPKB0424 remain unknown, both were found to influence bacterial growth, as their respective K/O mutants exhibited slower proliferation than the wild-type strain. Moreover, the reduced growth rates observed in all K/O mutants indicate that the differentially expressed genes identified in Hp52 contribute to bacterial colonization within the host (Supplementary Fig. 3). Previous studies have highlighted the importance of colonization efficiency in H. pylori infectivity (45, 46, 47, 48), yet the factors governing successful colonization remain poorly defined. Our results suggest that loss of any of these genes impairs colonization and proportionally reduces infectivity.
Dendritic cells (DCs) play a pivotal role in initiating and navigating immune responses to H. pylori, and therefore can be used in studies investigating H. pylori infection pathogenesis (24, 67). IFN-γ, although not the principal cytokine produced by bacteria-pulsed DCs, contributes to bacterial clearance (68, 69, 70, 71). In this study, DCs pulsed with any of the four K/O strains showed reduced IFN-γ expression compared with wild-type Hp52–pulsed DCs. In addition, pulsing with ΔHPKB0407, ΔHPKB0424, or Δald DCs resulted in decreased MHC class II expression, suggesting that these gene products function as important immunogenic antigens. Consistent with this, expression of the DC activation markers CD40 and CD80 was reduced in DCs stimulated with the K/O strains.
Interestingly, DCs exposed to ΔHPKB0346, ΔHPKB0407, and ΔHPKB0424 displayed reduced expression of IL-1β, IL-6, and IL-10, whereas Δald-pulsed DCs exhibited increased expression of these cytokines. These results imply that HPKB0346, HPKB0407, and HPKB0424 likely serve as major antigens that promote pro-inflammatory or Th17-type responses (27, 72, 73, 74, 75). Conversely, Ald appears to play a distinct immunomodulatory role, potentially promoting tolerogenic DC function. Given that H. pylori infection is known to shift the Th17/Treg balance toward immune tolerance (72), the increased IL-1β and IL-6 expression induced by the Δald mutant suggests that ald is involved in immune evasion mechanisms of H. pylori. In several bacterial pathogens, Ald has been implicated in metabolic adaptation under nutrient-limited or host-associated conditions, contributing to redox homeostasis and persistence (63, 64, 65). In H. pylori, ald-mediated metabolic regulation may similarly influence bacterial physiology in ways that indirectly modulate host immune responses. One possible mechanism is that Ald-dependent control of intracellular redox balance and amino acid metabolism affects the production or release of bacterial metabolites that act as immunomodulatory signals (76, 77). As mentioned above, Ald is a central metabolic enzyme that catalyzes the reversible conversion of L-alanine to pyruvate (62), and metabolic byproducts generated through alanine–pyruvate metabolism could influence host innate immune signaling pathways, including NF-κB activation and inflammasome-associated cytokines such as IL-1β (78, 79). Consistent with this notion, deletion of ald resulted in enhanced expression of IL-1β and IL-6 in dendritic cells, suggesting that Ald activity in the wild-type strain may normally dampen pro-inflammatory DC activation. However, it remains to be determined whether the increase in IL-10 expression in DCs pulsed by the Δald mutant represents a negative feedback response to heightened pro-inflammatory signaling such as increased IL-1β and IL-6 levels. Nevertheless, given the decreased IL-10 expression in DCs exposed to ΔHPKB0346, ΔHPKB0407, and ΔHPKB0424 mutants, it is plausible that the increased IL-10 expression in Δald-pulsed DCs is a result of negative feedback mechanisms responding to the elevated IL-1β and IL-6 levels (80, 81).
Through transcriptomic comparison and functional analysis of K/O strains, we identified four genes—ald, HPKB0346, HPKB0407, and HPKB0424—that contribute to bacterial growth rate and immune modulation. Notably, ald exhibited a unique role in altering DC immune responses, potentially facilitating chronic infection. Understanding the potential gene determinants that shape the immunological behavior of H. pylori may provide valuable insights for the development of vaccines or therapeutic strategies aimed at preventing or controlling H. pylori infection.