

INTRODUCTION
Nontuberculous mycobacteria (NTM) comprise all mycobacterial species other than Mycobacteriumtuberculosis (Mtb), M. africanum, M. bovis, M. canetti, M. caprae, M. pinnipedii, and M. leprae. These organisms are ubiquitous in soil and water, with approximately 200 identified species (1, 2). The M. avium complex (MAC) and the M. abscessus (Mabc) complex account for most cases of NTM pulmonary disease (NTM-PD). Both complexes cause disease in immunocompromised and apparently immunocompetent individuals (3). The Mabc complex comprises three subspecies: Mabc, M. abscessus subsp. massiliense, and subsp. bolletii. Subspecies abscessus and bolletii are frequently treatment-refractory because they carry erm(41), an inducible macrolide-resistance gene (4, 5). Despite the growing awareness of NTM-PD, the precise host–pathogen interactions that determine the outcome remain poorly understood, hindering therapy development. The rise of drug-resistant NTM infections therefore underscores an urgent need for novel and innovative therapeutic strategies to improve patient outcomes (6).
Autophagy is activated by metabolic, infectious, and inflammatory stress and targets impaired cellular organelles, intracellular proteins, and invading microbes by sequestering them in double- or multimembraned cytoplasmic vesicles called autophagosomes (7). These autophagosomes then fuse with lysosomes, where resident hydrolases degrade their cargo (8). Beyond its recycling role, autophagy safeguards cells against infection, inflammation, and immunometabolic imbalance (9, 10, 11). Recent studies have revealed that autophagy can act selectively, recognizing damaged organelles, protein aggregates, or intracellular bacteria to maintain intracellular homeostasis (12, 13). Selective autophagy relies on cargo receptors containing a microtubule-associated protein light chain 3 (LC3)-interacting region that bridge tagged substrates to the autophagic machinery (12, 13). In this Review, we briefly discussed bulk and selective autophagy and host–pathogen interactions during NTM infection. We further examined how autophagy-related pathways and genes influence host defense and susceptibility to NTM as well as highlighted agents that activate host autophagy and lysosomal function to enhance antimicrobial immunity.
OVERVIEW OF AUTOPHAGY
Autophagy classification
Autophagy is broadly classified into macroautophagy (hereafter referred to as autophagy), microautophagy, and chaperone-mediated autophagy. Microautophagy directly sequesters cellular material—including KFERQ-tagged proteins and bulk cytosol—via membrane invaginations or protrusions of late endosomes or lysosomes, in an endosomal sorting complex required for transport (ESCRT)-dependent or -independent manner (14, 15, 16, 17, 18). Unlike macroautophagy, microautophagy bypasses autophagosome formation and is usually non-selective, although specialized selective variants have been identified (19). Chaperone-mediated autophagy targets specific proteins containing KFERQ-like motifs via the cytosolic chaperone, heat shock cognate protein HSC70. HSC70 delivers these substrates to the lysosomal membrane receptor, lysosomal associated membrane protein (LAMP) 2A, which mediates their translocation into the lysosomal membrane for degradation (20).
Autophagy process
Autophagy, including its selective forms, is mediated by autophagy-related genes (ATGs), which regulate every pathway stage (Fig. 1). ATGs control the initiation, elongation, and maturation of autophagosomes and enable cells to adapt to stress or nutrient deprivation (21). Autophagy is initiated when the UNC-51-like kinase (ULK) complex—comprising focal adhesion kinase family interacting protein of 200 kDa (FIP200), ATG13, ATG101, and serine/threonine kinases ULK1 and ULK2—is activated, a step normally inhibited by mammalian target of rapamycin complex 1 (mTORC1) (22, 23, 24). During energy depletion, AMP-activated protein kinase (AMPK) inactivates mTORC1, thereby permitting ULK-complex assembly at the endoplasmic reticulum membrane and recruitment of downstream ATGs that induce autophagosome formation (25). Next, the ULK-complex recruits class III phosphatidylinositol-3 (PI(3))-kinase complex 1 (PI3KC3-C1), another critical complex for autophagy initiation, to produce PI(3) phosphate (P) on the autophagic membrane. PI3KC3-C1 comprises vacuolar protein sorting (VPS) 34, VPS15, Beclin-1 (BECN1), ATG14, and nuclear receptor binding factor 2 (NRBF2) (26). PI3KC3-C1 binds to membranes via ATG14, BECN1, and VPS34, subsequently catalyzing PI(3)P production by VPS34, thereby facilitating omegasome formation (26, 27). Subsequent autophagosome biogenesis relies on WD-repeat protein interacting with phosphoinositides proteins—effectors of PI(3)P—and two ubiquitin-like conjugation systems (28, 29). The latter comprise (i) the ATG12–ATG5–ATG16L1 complex (assembled by ATG7 and ATG10) and (ii) the cascade that lipidates ATG8 family proteins via ATG7 and ATG3. Lipidated ATG8 family members are essential for phagophore expansion and cargo recognition. The cysteine protease ATG4 cleaves pro-ATG8s at their C-terminus, exposing the glycine residue required for conjugation to phosphatidylethanolamine (PE) (30). Among the four mammalian ATG4 isoforms, ATG4A preferentially processes gamma-aminobutyric acid receptor-associated proteins, whereas ATG4B has broad specificity for all ATG8s (31, 32). Processed ATG8s are then activated by the E1-like enzyme ATG7 and transferred to membrane-bound PE by ATG3 (8). Consequently, soluble ATG8s (e.g., LC3-I) are converted into membrane-anchored, lipidated forms (e.g., LC3-II) (28, 33). WD-repeat protein interacting with phosphoinositides 2 recruits the ATG12–ATG5–ATG16L1 complex to phagophores, where it acts as an E3-ligase for ATG8 lipidation, drives phagophore expansion and maturation, and ultimately promotes autophagosome–lysosome fusion (28).
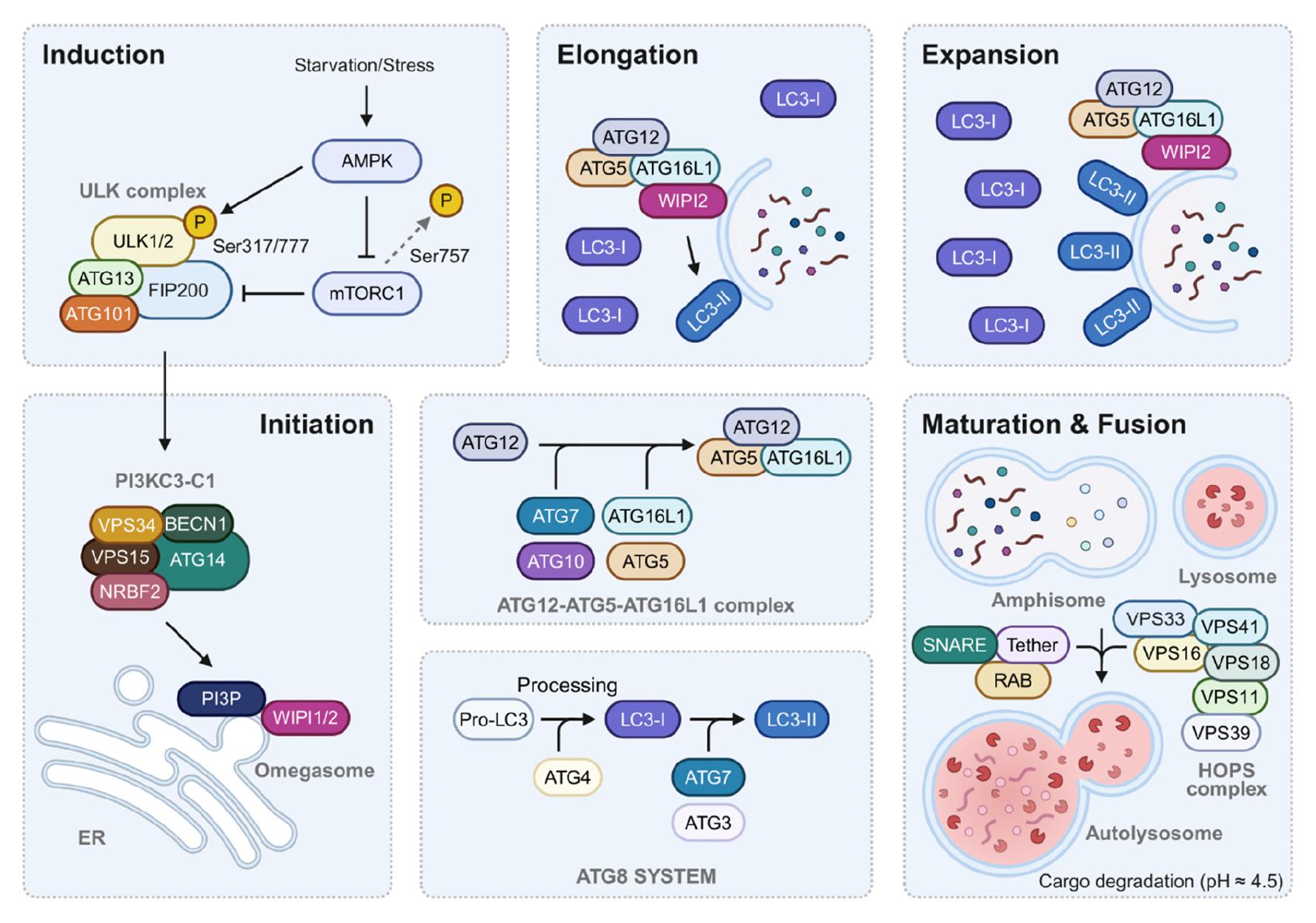
Fig. 1
Schematic representation of the macroautophagy pathway and its key molecular components. The macroautophagy pathway proceeds through distinct stages: induction, initiation, elongation, and maturation/fusion. The process is initiated by stress signals or starvation, activating the ULK complex (ULK1/2, ATG13, FIP200, and ATG101) and its regulation by mTORC1 and AMPK. The initiation phase involves the PI3KC3-C1 complex (containing VPS34, BECN1, VPS15, ATG14, and NRBF2) which generates PI(3)P at the omegasome. During elongation, two ubiquitin-like conjugation systems operate: the ATG12-ATG5-ATG16L1 complex and the ATG8/LC3 system. LC3 undergoes processing from pro-LC3 to LC3-I and finally to membrane-bound LC3-II. The maturation phase involves fusion with lysosomes, facilitated by the HOPS complex, RAB proteins, and SNARE complexes, ultimately leading to cargo degradation in the acidic autolysosome (pH ≈ 4.5). P, phosphorylation; ER, endoplasmic reticulum; ATG, autophagy-related genes; HOPS, homotypic fusion and vacuole protein sorting complex; LC3, microtubule-associated protein 1 light chain 3; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptors.
Autophagy maturation involves the fusion of autophagosomes with vesicles from endolysosomal compartments to form amphisomes, leading to the formation of degradative autolysosomes. This process is regulated by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) proteins, ATG8 family members, membrane tethering factors, and Rab GTPases (34, 35). Several regulatory mechanisms underlying autophagosome maturation are currently being identified. These mechanisms are modulated by post-translational modifications of key proteins, spatial–temporal regulation of phosphoinositide lipids, Rab GTPase activity, and lysosome biosynthesis and degradation (34, 35). Impaired autophagosome maturation is associated with the pathogenesis of various human diseases, including neurodegeneration, cancer, and lysosomal storage disorders (34, 35). Further research into autophagy maturation will enhance our understanding of disease mechanisms and support the development of novel therapeutics against infection.
Selective autophagy: xenophagy
Selective autophagy targets specific cellular components—such as damaged organelles (e.g., mitophagy and pexophagy), protein aggregates (aggrephagy), and intracellular bacteria (xenophagy)—for lysosomal degradation. This process begins when autophagy receptors possessing ATG8-interacting motifs (AIMs) or LC3-interacting regions (LIRs) recognize and bind specific cargo. These receptors interact with ATG8 family proteins, such as LC3 and gamma-aminobutyric acid receptor-associated proteins, facilitating cargo delivery to the autophagy machinery and eventual lysosomal degradation (Fig. 2) (12, 13, 36). Xenophagy, a subtype of selective autophagy, targets intracellular pathogens, such as Mycobacterium, Salmonella, and Listeria, by tagging them with ubiquitin. Ubiquitinated pathogens are recognized by selective autophagy receptors—including p62 (sequestome-1), neighbor of BRCA1 gene 1 (NBR1), optineurin (OPTN), and nuclear dot protein 52 kDa (NDP52)—which link them to autophagy machinery for lysosomal degradation (13, 37). However, several intracellular pathogens have evolved mechanisms to disrupt key xenophagy steps—including ubiquitination, cargo recognition, autophagosome formation, or lysosomal fusion—thus evading degradation. For example, Mtb impairs autophagosome acidification, whereas Salmonella secretes effectors that disrupt host autophagy signaling to escape lysosomal degradation (38, 39, 40). Additionally, some pathogens manipulate autophagy-related pathways to establish replication- permissive niches during chronic infection (41). Extensive reviews (42, 43, 44, 45, 46, 47, 48, 49) have examined pathogen strategies to modulate host autophagy pathways. Therefore, this Review does not detail individual pathogen strategies to subvert host autophagy or xenophagy during infection.

Fig. 2
Xenophagy pathways. Xenophagy involves these five steps: (1) Phagocytosis and escape from the phagosome or membrane damage. (2) Bacteria tagged with ubiquitin molecules. (3) Autophagic receptors like p62, NBR1, OPTN, and NDP52 recognize tagged bacteria. (4) Autophagosome formation. (5) Lysosomal fusion and degradation. Autophagic receptors have two key domains: UBD and AIM/LIR. They link ubiquitinated bacteria and LC3 on autophagosomal membrane, leading to sequestration of bacteria within the autophagosome. Ub, Ubiquitin molecule; UBD, Ubiquitin binding domain; AIM/LIR, Atg8-interacting motif/LC3-interacting region; OPTN, optineurin; NDP52, nuclear dot protein 52 kDa; NBR1, neighbor of BRCA1 gene 1; LC3, microtubule-associated protein 1 light chain 3.
Antibiotic resistance has become a critical global health concern. Multiple NTM species are notoriously difficult to treat because of their intrinsic drug resistance (50). To address NTM drug resistance, researchers are exploring novel host- directed approaches—including autophagy-inducing nanoformulations and siRNA-based gene silencing—alongside chimeric antibiotics designed to overcome multidrug-resistant infection (39). Additionally, animal infection models suggest that several autophagy proteins function beyond xenophagy to influence bacterial pathogenesis, suggesting that pathogens broadly modulate host surveillance pathways (38). Understanding how bacteria evade xenophagy and how pathogens interfere with host autophagy is crucial for developing advanced therapies that restore effective immunity to intracellular pathogens, including NTMs (38, 39).
NTM INFECTION AND HOST IMMUNE DEFENSE MECHANISMS
Although generally environmental and non-pathogenic, NTMs can cause various infections in the lungs, lymph nodes, central nervous system, and skin (51). NTM-PD is the most prevalent presentation and is increasing globally in both immunodeficient and immunocompetent individuals (52). Macrophage-driven innate immunity orchestrates the initial host defense and primes adaptive responses during NTM infection (52). Innate immune receptors and their signaling cascades induce the activation of antimicrobial proteins and inflammatory mediators in the innate immune network during infection (53, 54). Both hyper- and hypo-inflammatory states can compromise host defense and drive pathological progression (53). Autophagy is a well-known effector system of innate immunity (54), but its specific role in NTM infection remains underexplored. Additionally, how NTMs evade autophagic defense is poorly understood. The next section comprehensively discusses current insights into autophagy during NTM infection.
Additionally, NTM successfully subvert adaptive immunity, promoting virulence and persistent infection even after antimicrobial chemotherapy (51, 55). NTM infection contributes to T-cell exhaustion, characterized by diminished interferon (IFN)-γ production and continuous expression of inhibitory receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programed cell death protein 1 (PD-1), and T-cell immuno- globulin domain and mucin domain 3 (TIM-3) (56). Elucidating immune dysregulation and exhaustion should reveal key prognostic, predictive, and therapeutic biomarkers that reflect—and potentially recalibrate—the patient’s immune status. Such knowledge could inform host-directed immunomodulatory therapies that restore T-cell function and maintain inflammatory homeostasis, enabling more personalized treatment.
Individual NTM species employ diverse strategies to survive and replicate intracellularly, subverting multiple host-defense pathways, yet the specific microbial effectors that drive immune evasion remain poorly defined. Among NTM species, Mabc enhances cellular survival by inducing phagosome pores, suppressing phagosome maturation, modulating inflammatory cytokine production, and delaying host cell apoptosis (57). The ESX-4 (one of type VII) secretion system is central to this process: deletion of the key gene eccB4 reduces Mabc’s capacity to prevent phagosomal acidification and membrane damage, highlighting the critical role of ESX-4 in Mabc virulence (58). M. avium (Mav) causes chronic lung infection in mice, residing in granuloma-like structures that shield it from immune clearance (59). Mav control depends on coordinated innate (e.g., lipocalin 2 and neutrophils) and adaptive (e.g., IFN-γ and antibodies) responses (59). In human primary macrophages, mitochondrial import of pyruvate has recently been shown to enhance reactive oxygen species (ROS) via reverse electron transport at complex I, restricting Mav growth (60). Exploring the detailed mechanisms underlying these host–NTM interactions should facilitate the development of immunotherapeutic strategies to correct immune dysfunction and combat chronic infection.
NTM INFECTION AND AUTOPHAGY
Autophagy during NTM infection
An early study reported that therapeutic concentrations of azithromycin suppress lysosomal acidification in human primary macrophages, thereby impairing the autophagic clearance of Mabc (61), underscoring the importance of autophagy in NTM defense. A recent study showed that host macrophage effector responses vary between NTM species. In human THP-1 macrophages, autophagosome formation is greater after infection with M. abscessus and M. smegmatis than with M. intracellulare, Mav, or Mtb (62). Pathogenic potential also differs between smooth (S) and rough (R) variants of Mabc (Mabc-S and -R) (63). Mabc-S persists intracellularly by blocking phagosome maturation and allowing limited phagosome–cytoplasm communication. In contrast, Mabc-R induces stronger autophagy and apoptosis than Mabc-S and forms extracellular cords (63, 64). These findings highlight the need for therapies targeting both intracellular bacteria and extracellular aggregates (63).
During infection, M. marinum becomes ubiquitinated in host macrophages and is sequestered in LAMP-1-positive vacuoles resembling lysosomes. Interestingly, this process occurs independently of classical autophagy (Fig. 3). ESX-1 secretion is required for M. marinum escape into the cytosol but not for its ubiquitination (65). These findings suggest that ubiquitinated M. marinum is targeted for lysosomal degradation via a non-canonical, autophagy-independent pathway or escape it to remain cytosolic and exploit actin-based motility (65). Moreover, a previous study used a zebrafish model to analyze the autophagy response to infection at high resolution in vivo. During infection, M. marinum is primarily associated with green fluorescent protein (GFP)-LC3-positive vesicles, and ~70% of larger autophagic vesicles are colocalized with lysosomal markers, mainly in leukocytes (66). This in vivo platform offers dynamic insights into autophagy-mediated defense against diverse NTM species.
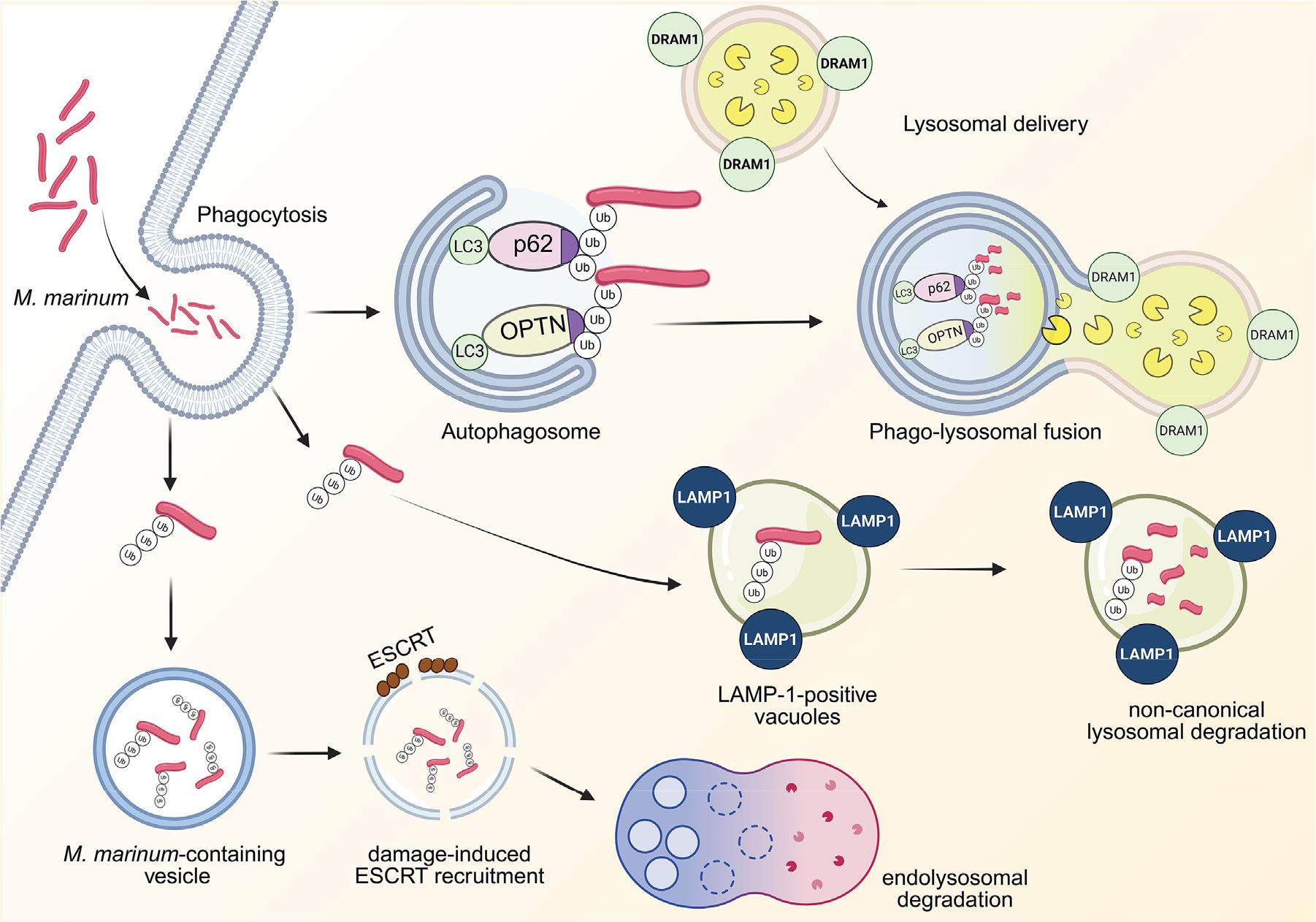
Fig. 3
Autophagy during M. marinum infection. M. marinum is phagocytosed, activating its ubiquitination. The ubiquitinated M. marinum is eliminated by LAMP-1-positive vacuoles via non-canonical lysosomal degradation. The ubiquitinated M. marinum is captured by p62 and OPTN, which navigate toward the LC3-conjugated autophagosome. DRAM1 promotes the lysosomal delivery to the autophagosome, leading to the phagolysosomal fusion. The ESCRT is recruited to the damaged vesicles containing ubiquitinated M. marinum, eliciting the endolysosomal degradation. DRAM1, DNA damage-regulated autophagy modulator 1; LC3, microtubule-associated protein 1 light chain 3; OPTN, optineurin; Ub, ubiquitin; LAMP1, lysosome-associated membrane protein 1; ESCRT, endosomal sorting complex required for transport.
Selective xenophagy receptors, OPTN, and p62 are essential for host defense against M. marinum, as shown in the zebrafish model (67, 68). OPTN and p62 compensate for each other’s loss of function and act additively to protect against M. marinum infection; zebrafish lacking both receptors showed greater susceptibility than either single mutant (67). Damage-regulated autophagy modulator-1 (DRAM1) interacts with M. marinum in macrophages during infection. DRAM1-positive bacilli colocalize with autophagosomes and lysosomes, promoting lysosomal delivery and enhancing macrophage bactericidal activity (69). DRAM1 is required for phagosome maturation, and its deficiency exacerbates pyroptotic cell death during M. marinum infection (Fig. 3) (70). Notably, both OPTN and p62 still confer protection in DRAM1-deficient zebrafish, indicating that xenophagy receptors enhance antimycobacterial defense independently of DRAM1 (67). Additionally, in the soil ameba Dictyostelium discoideum, infectionrecruits ESCRT proteins tumor susceptibility gene 101, VPS32, and VPS4 to damaged regions of the pathogen-containing vacuole (Fig. 3) (71). The TNF receptor-associated factor (TRAF)-like E3 ligase TrafE is essential for the ESCRT- and autophagy-mediated endolysosomal damage response that restricts M. marinum via xenophagy in D. discoideum(72). Future studies are needed to elucidate how selective autophagy receptors and the related ESCRT system activate host defense against diverse NTM species and identify small molecules or drugs that selectively boost xenophagy as autophagy-based therapeutics against NTM infection.
ATGs and NTM infection
In the Drosophila infection model, M. marinum activates cytokine–signal transducer and activator of transcription signaling, inhibits Atg2 expression, and drives abnormal lipid-droplet accumulation, creating a better environment for bacterial survival (Fig. 4) (73). Similarly, interleukin-6 (IL-6) enhances mycobacterial survival and lipid deposition in human macrophages (73). These studies suggest that augmenting key autophagy genes or restoring lipid homeostasis could restrict NTM survival within host cells, but elucidating the precise mechanisms requires further study.

Fig. 4
ATGs and NTM infection. M. marinum infection activates cytokine-STAT3 signaling pathways, leading to the upregulation of Atg2 and the abnormal accumulation of lipid droplets. It suppresses the survival of M. marinum within host system. Under mycobacteria-containing vacuole conditions, M. marinum ESX-1 promotes the expression of Atgs, leading to the formation of autophagosome. However, M. marinum ESX-1 inhibits autophagic flux. Additionally, M. marinum MimG, an ortholog of Mtb Rv3242c, suppresses autophagic process, thus eliciting an increase in bacterial survival. M. smegmatis is recognized by TLR2 and activates the transcription of multiple ATGs, resulting in the induction of alternative autophagy, which is independent of ubiquitination or autophagy receptors. This pathway ultimately reduces bacterial survival. The secretion of mycolactone toxin by M. ulcerans triggers necrosis, thereby increasing bacterial evasion. STAT3, signal transducer and activator of transcription 3; ATG, autophagy-related gene; TLR2, toll-like receptor 2.
Transcriptomic analyses revealed that M. marinum—but not Legionella pneumophila—activates autophagy- and ESCRT- related genes in D. discoideum, pointing to pathogen-specific activation of these pathways (74). The non-pathogenic M. smegmatis triggers a non-canonical autophagy pathway in THP-1 macrophages toll-like receptor 2 (TLR2) and ATG upregulation; LC3 recruitment is ubiquitin- and receptor-independent yet still suppresses intracellular M. smegmatis survival (Fig. 4) (75). The ESX-1 secretion system of M. marinum is required to both upregulate autophagy genes and manipulate the pathway. M. marinum ESX-1 recruits autophagosomes to the mycobacteria-containing vacuole (MCV) where the bacterium survives (Fig. 4). Simultaneously, ESX-1 suppresses autophagic flux and cargo degradation within the MCV, thereby favoring bacterial survival inside host cells (76). A previous study showed that mycolactone, the exotoxin produced by M. ulcerans—the causative agent of Buruli ulcer—induces necrosis, enabling bacterial evasion of host immunity (Fig. 4). Notably, mycolactone also activates mTOR signaling and suppresses autophagy in infected macrophages (77). Additionally, Mtb Rv3242c—encoding A mannosylated glycoprotein phosphoribosyltransferase—and its M. marinum ortholog MimG suppress autophagy, thereby preventing intracellular bacterial clearance (Fig. 4) (78). Collectively, these findings suggest that various NTM effectors act as virulence and immunomodulatory factors by suppressing host autophagy, highlighting their potential as novel therapeutic targets. Consequently, detailed studies are warranted to determine how individual NTM species and their effectors drive pathogenesis and evade host defense mechanisms, including autophagy, to inform preventive and therapeutic development against NTM infections.
Specific genetic variants of ATGs and the risk of NTM infection
There is growing evidence suggesting that autophagy genes influence host protection against, or susceptibility to, NTM infection. A genome-wide association study of Buruli ulcer patients and controls showed that the protective G allele of an ATG16L1 missense variant correlates with decreased antibacterial autophagy, implicating autophagy in disease development (79). Additionally, variants in PARK2 (rs1333955) and NOD2 (rs9302752 and rs2066842) are associated with an increased risk of Buruli ulcer, whereas ATG16L1 rs2241880 appears to protect against the ulcerative form of Buruli ulcer (80). Furthermore, in Korean patients with NTM-PD, PARK2 expression is downregulated and linked to host susceptibility (81). These findings suggest that genetic variation in ATGs influences both the risk and progression of NTM disease.
AUTOPHAGY-TARGETING STRATEGIES AGAINST NTM INFECTION
Although most NTM species are environmentally non-pathogenic, human infections are rising worldwide and are difficult to treat due to intrinsic resistance to commonly used antibiotics (50). Several agents that do not directly kill NTMs nevertheless enhance host defense against these pathogens in vitro and in vivo. This section discusses recent findings that autophagy mediates the host-directed antimicrobial effects of such compounds during NTM infection (Table 1). An early study showed that 1,25(OH)₂D₃ and LL-37—the C-terminal peptide of CAMP—promote autophagy-mediated killing of M. marinum in THP-1 cells (82). Together with findings in vitamin-D-treated Mtb-infected macrophages (83), these data strongly indicate that activation of the vitamin D receptor—CAMP axis enhances autophagy-dependent defense in human monocytic cells.
Table 1.
Therapeutics targeting autophagy against NTM infection
| Drug candidate | Infectious agent | Mechanism of action | Study model | Biological actions | Ref |
|---|---|---|---|---|---|
|
1, 25-Dihydroxyvitamin D3 | M. marinum | Induction of CAMP |
THP-1, U937, Atg5−/− MEF | Induced CAMP improved the antimicrobial activity via autophagolysosome localization and suppressed pro-inflammatory cytokines | (82) |
| Trehalose |
MAC M. fortuitum | Activate PIKFYVE, TFEB nuclear translocation | U937, PBMC from healthy or HIV infected donors, mouse | Induced HIV-mediated inhibition of xenophagy flux, reduced HIV-P24 levels and restricted NTM survival during single or HIV co-infections | (84) |
| 2-DG | M. marinum | Autophagy induction |
RAW264.7, WT and Tnf−/− zebrafish | Decreased phagocytosis and restricted M. marinum growth by autophagy induction and enhancing TNF-α expression | (85) |
|
Trifluoperazine, Chlorproethazine | M. avium | TFEB nuclear translocation | Purified CD14+ monocytes from healthy donors | Restricted Mav survival via reduced NADPH oxidase-driven ROS and mildly enhanced autophagy flux | (86) |
| Dimethyl itaconate | M. avium | Autophagy flux activation |
Macrophages from Atg7fl/fl, Atg7−/− mice, BMDMs, and PMs, in vivo mouse model | Enhanced autophagy and phagosomal maturation are associated with reduced levels of IL-6/IL-10 and STAT3, autophagy partially contributed to the antimicrobial response. | (87) |
| V46 | M. abscessus, MDR-M. abscessus | TFEB nuclear translocation | BMDMs, macrophages from Atg7fl/fl and Atg7−/− mice, mouse | Enhanced bacterial clearance in macrophages and lungs via autophagy activation, Inhibited pro-inflammatory cytokines and chemokines | (89) |
| Amiodarone | M. avium | TFEB nuclear translocation | Human macrophages, zebrafish embryo | Reduced bacterial burden via TFEB-regulated autophagy and lysosomal biogenesis in macrophages and zebrafish embryos | (90) |
| Rufomycin | M. abscessus, rough strain of M. massilense | TFEB nuclear translocation | BMDMs, mouse | Facilitated bacterial eradication by augmenting autophagy and lysosomal gene expression, decreased mitochondrial dysfunction and oxidative stress | (91) |
| Degarelix | M. marinum | PI3Kinase inhibition | BMDMs, mouse and zebrafish | Inhibited bacterial burden and necrotic granuloma fraction, elevated IFN-γ expression levels in M. marinum infected zebrafish | (92) |
| Raloxifene | M. abscessus | Autophagy activation | A549, huh7, RAW264.7 | Prevented bacterial invasion and proliferation by enhancing TRIM/ GABA pathways, inhibited cholesterol biosynthesis identified via transcriptomic profiling | (93) |
| TSR | M. marinum, | ER stress-mediated autophagy |
RAW264.7, zebrafish larvae | Reduced intracellular bacterial load by promoting the ER stress pathway through eIF2α and PERK phosphorylation and enhanced LC3-II conversion | (94) |
| Rapamycin | M. smegmatis | Non-autophagy pathway | RAW264.7, A549, LC3B and Atg5-deficient macrophages | Antimicrobial effects are seen at higher concentration, independent on LC3B and Atg5 related pathways | (95) |
| AAT | M. intracellulare | Autophagy activation | PBMC from AAT-infused patients, human alveolar macrophages, THP-1 | Reduced bacterial burden by enhancing phagosome-lysosome fusion, autophagosome maturation, and inhibiting NF-κB and A20 expression | (96) |
| IL-17A, IL-17F | M. terrae | Autophagy activation | RAW 264.7 | IL-17F reduced the intracellular bacterial load by increasing the number and size of autophagosomes and LC3B-II accumulation, IL-17F stimulated autophagy more effectively than IL-17A | (97) |
| Metformin | M. avium | AMPK activation | hMDMs, BMDMs, mouse | Increased the number of T cells that produce IFN-γ in lung thus limiting Mav growth, elevated mtROS production, phagosomal maturation, and acidification in macrophages | (98) |
AAT, alpha-1-antitrypsin; BMDMs, bone marrow derived macrophages; CAMP, cathelicidin antimicrobial peptide; 2-DG, 2-deoxyglucose; hMDMs, human monocyte-derived macrophages; HIV, human immunodeficiency virus; MAC, Mycobacterium avium complex; MDR, multidrug-resistant; PI3 Kinase, Phosphoinositide 3-kinase; PIKFYVE, a FYVE finger-containing phosphoinositide kinase; PM, peritoneal macrophages; TSR Thiostrepton
Trehalose, a disaccharide and potent autophagy inducer, induces aggrephagy and suppresses Mtb and NTM growth in human macrophages, including during co-infection with HIV (84). Mechanistically, trehalose activates PIKFYVE–transcription factor EB (TFEB) signaling and enhances xenophagic flux, thereby restricting mycobacterial replication (84). These reports suggest that there are therapeutic strategies that boost autophagy and lysosomal function for NTM infections, whether isolated or concurrent with HIV. Pretreatment of zebrafish larvae with 2-deoxy-D-glucose, a glycolysis inhibitor, confers protection against M. marinum, possibly via increased TNF-α production (85). These findings suggest that the time point to treat NTM infection is essential for aerobic glycolysis-associated host protection. Future studies should determine how manipulating aerobic glycolysis influence infection outcome and assess whether such modulators can serve as preventive treatments or vaccine adjuvants for NTM infection.
Phenothiazines—specifically trifluoperazine and chlorproethazine—suppress intracellular Mav growth in primary human macrophages and increase autophagic flux and nuclear translocation of TFEB (86). However, this autophagy activation does not appear to drive their host-defense effect. Instead, partial control of Mav involves inhibition of NADPH oxidase- mediated ROS production (86). Dimethyl itaconate (DMI)—a cell-permeable ester derivative of itaconate—exerts multifaceted host-defense effects against Mtb and Mav, at least partly via autophagy activation (87). Signal transducer and activator of transcription 3 (STAT3) regulates autophagy transcriptionally by controlling ATGs such as BECN1, PIK3C3, cathepsin B/L, and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 and can suppress autophagy through interactions with forkhead box O 1 and 3 (88). Because DMI inhibits STAT3 signaling during mycobacterial infection (87), it will be important to determine whether this STAT3 inhibition further enhances—rather than blocks—autophagy-mediated host defense.
Recent studies suggest that TFEB is a promising target for host-directed therapeutics against NTM infection. V46, a resveratrol analog, significantly restricts the growth of both S and R Mabc morphotypes—including multidrug-resistant strains—in macrophages and in mouse lungs. Its activity partly involves reduced pro-inflammatory cytokines/chemokines and enhanced autophagy with TFEB nuclear translocation during Mabc infection (89). Despite being a resveratrol analog, V46 acts independently of SIRT1 or SIRT3 expression (89). Amiodarone—an anti-arrhythmic drug—exerts therapeutic effects against Mtb and Mav in primary human macrophages by activating autophagy and directing mycobacteria to autophagosomes (90). Amiodarone-induced autophagy correlates with increased TFEB activity and nuclear translocation, and blocking autophagic flux with bafilomycin diminishes its antimicrobial effect (90). Rufomycins 4–7, which target mycobacterial ATP-dependent Clp protease ATP-binding subunit ClpC1, exert dual antimicrobial actions against R strains of Mabc. Host-side, they suppress inflammation and promote TFEB nuclear translocation, thereby upregulating autophagy and lysosomal genes (91). Together, these data strongly suggest that TFEB activation is critical for the suppression of NTM infection. Future studies should elucidate how TFEB and related transcription factors can be harnessed to combat NTM disease.
The anticancer drug degarelix, despite lacking direct antimycobacterial activity, significantly reduces intracellular Mtb H37Rv survival and suppresses pathology in M. marinum-infected zebrafish models(92). This effect depends, at least in part, on the autophagy-initiation pathway (92). Raloxifene, a broad-spectrum antibacterial drug, transcriptionally upregulates autophagy pathways—including TRIM and GABA signaling—thereby increasing autophagy and preventing bacterial invasion and proliferation during methicillin-resistant Staphylococcus aureus and Mabc infection (93). Derivatives of thiostrepton, a thiopeptide antibiotic that targets bacterial ribosomes, induce ER stress-mediated autophagy in host cells and enhance intracellular elimination of M. marinum(94). These findings suggest that various drugs and their derivatives can boost host defense by stimulating autophagy, offering new insights for NTM therapy (94). Conversely, rapamycin an mTOR inhibitor, shows antibacterial activity against M. smegmatis only at high concentrations and even in LC3B- and ATG5-deficient macrophages (95). Thus, autophagy activators may exert context-dependent antimicrobial effects against NTM through mechanisms beyond canonical autophagy activation. Future studies should elucidate the autophagic and non-autophagic mechanisms of these pharmacological models at therapeutically effective concentrations.
Infusion with α1-antitrypsin (AAT) augments host defense against M. intracellulare in monocyte-derived macrophages and in plasma. In macrophages, AAT significantly increases phagolysosomal fusion and autophagy activity. AAT-induced autophagy is mediated by inhibition of M. intracellulare-induced NF-κB activation and A20 expression (96). Both IL-17A and IL-17F enhance autophagic flux, increase autophagosome number and size, and promote the formation of acidic vesicular organelles in RAW264.7 cells. IL-17F is more effective than IL-17A and significantly reduces intracellular M. terrae growth, suggesting that IL-17-mediated autophagy contributes to host defense during NTM infection (97). Furthermore, metformin treatment significantly reduces the pulmonary bacterial load in Mav-infected mice and increases the lung infiltration of Mav-specific IFN-γ-producing T cells (98). Given earlier findings that metformin activates AMPK-dependent autophagy (99), future studies should determine whether its efficacy against Mav lung disease depends on boosting macrophage autophagy.
CONCLUSIONS
Given that NTM pulmonary infections are difficult to treat—owing to multidrug resistance and chronic persistence—interest in host-directed therapy is increasing. Understanding the roles and mechanisms influencing autophagy activation and lysosomal biogenesis may contribute to the development of novel, effective therapies for NTM pulmonary disease. Research on autophagy-mediated host defense against NTM infection is advancing rapidly: recent studies have identified novel molecules and pathways that activate xenophagy and improve the host defense system against NTMs. Despite this, it remains largely unknown how individual NTM species and their effectors evade, manipulate, or disrupt host autophagy to enhance their pathogenesis and virulence. Many key questions remain: Which ATGs and pathways are most critical for controlling NTM infection in vivo? How do pathogen-specific evasion mechanisms operate, and how can we target them therapeutically? Additional research is warranted to understand the molecular interactions of selective autophagy types—mitophagy, xenophagy, and others—interact with other biological pathways and immune responses during NTM infection. Such insights will clarify the nuanced regulatory mechanisms that determine NTM infection outcomes.
Emerging evidence points to autophagy-activating compounds as adjuncts that boost host immunity and enhance conventional antibiotic efficacy. This host-directed approach could open new avenues for the treatment of NTM infection by reinforcing immunity and overcoming pathogen resistance. However, their clinical usefulness remains to be defined. The efficacy and safety of autophagy inducers in clinical settings remain uncertain. Well-designed preclinical and clinical trials are necessary to evaluate these agents, accounting for NTM pathophysiology and patient immune status. These efforts may elucidate the management of NTM disease using autophagy-targeted therapy.
