

INTRODUCTION
Vibrio vulnificus is a halophilic bacterium commonly found in estuarine environments, known to cause opportunistic infections in susceptible hosts following the consumption of contaminated seafood or through wound infections, often resulting in a mortality rate exceeding 50% (1, 2, 3). Upon infecting hosts, V. vulnificus undergoes profound environmental shifts from its natural habitat to human hosts, as evidenced by our previous identification of in vivo-induced genes using the in vivo-induced antigen technology (IVIAT) (1). Intriguingly, computational simulations employing the genome-scale metabolic network model VvuMBEL943 have revealed discrepancies, with specific gene clusters showing heightened metabolic fluxes despite lacking in vivo up-regulation (4). For instance, the ATP synthase Ι cluster (Vv1_1014 to Vv1_1022) exhibited significant increases in metabolic fluxes during in vivo analyses. Our investigations have further unveiled extensive metabolic reprogramming in V. vulnificus post-infection, with over 150 genes identified as up-regulated in vivo based on genome-based analyses (5). Notably, among these genes, the NADH dehydrogenase gene (Vv1_2074) has emerged as a significant candidate. This inconsistency prompted us to hypothesize that post-infection, the enzymatic activity of ATP synthase Ι and the expression of NADH dehydrogenase are critical for V. vulnificus adaptation within the host milieu.
To validate these hypotheses, we previously characterized the ATP synthase Ι gene cluster and observed in vivo growth retardation and reduced intracellular ATP levels in insertion mutants under O2-limited conditions (unpublished data). This suggests the pivotal role of ATP synthase Ι in energy production, particularly during V. vulnificus infection in vivo. Subsequently, our attention turned to the origin of protons essential for ATP synthesis via ATP synthase Ι. Typically, in cellular respiration, five membrane-bound complexes (complexes I-V) work together to facilitate electron transport and oxidative phosphorylation, thereby supplying ATP synthase Ι (complex V) with the requisite charge for energy production (6, 7, 8, 9). However, our systems biological analysis uncovered a distinctive mechanism for ATP generation in vivo. Specifically, we observed up-regulation of only complex I within the membrane-bound electron transport chain, while complexes II, III, and IV were down-regulated (5).
NADH dehydrogenase, often referred to as complex I, facilitates the coupling of electron movement to proton transfer, thereby converting energy. Structural analyses of complex I have revealed an electron transfer mechanism that drives the pumping of four protons (4H+) from the cytoplasm across the cell membrane. This proton gradient is then utilized to generate ATP molecules through complex V on the extracellular membrane (8, 10), we postulate that it serves as the primary proton source for ATP synthesis via ATP synthase Ι post-infection. This study aims to characterize the role of NADH dehydrogenase in V. vulnificus post-infection energy production by constructing deletion mutants and revertant strains and assessing their characteristics under both in vitro and in vivo growth conditions.
MATERIALS AND METHODS
Bacterial strains, plasmids and culture media
Bacterial strains and plasmids utilized in this study are detailed in Table 1. Vibrio vulnificus CMCP6, obtained from a male patient admitted to Chonnam National University Hospital, South Korea, serves as a clinical isolate. V. vulnificus CMCP6 was cultivated in 2.5% NaCl heart infusion (HI) medium, while Escherichia coli strains were cultured in Luria-Bertani (LB) medium supplemented with appropriate antibiotics. Cultures were maintained at 37°C with agitation at 200 rpm. Thiosulfate citrate bile salt sucrose agar (TCBS; Merck, Darmstadt, Germany) was employed as the selective medium for V. vulnificus. Antibiotic concentrations for E. coli were as follows: ampicillin (Amp) 100 μg/mL, kanamycin (Km) 100 μg/mL, chloramphenicol (Cm) 30 μg/mL, and tetracycline (Tc) 12.5 μg/mL. For V. vulnificus, ampicillin (20 μg/mL), tetracycline (2 μg/mL), and chloramphenicol (2 μg/mL) were used.
Table 1.
Bacterial strains and plasmids used in this study. Apr, Ap resistance; Kmr, Km resistance; Tcr, Tc resistance; Cmr, Cm resistance
| Strains or plasmids | Description | Source or reference |
|---|---|---|
| Vibrio vulnificus | ||
| CMCP6 | Clinical isolate | (47) |
| ΔVV1_2074 | CMCP6 with Vv1_2074 deletion mutant of complex I | This study |
| ΔVV1_2074 revertant | Revertant strain of ΔVV1_2074 after replacement of VV1_2074 gene | This study |
| Escherichia coli | ||
| DH5α | F—recA1; restriction negative | Laboratory collection |
| SY327λpir | Δ (lac pro) argE(Am) rif nalA recA56 λ pir lysogen; host for π-requiring plasmids | (48) |
| SM10 λpir | thi thr leu tonA lacY supE recA::RP4-2-TcR::Mu λ pir lysogen, oriT of RP4, KmR ; Conjugal donor | (48) |
| Plasmids | ||
| pCR2.1TOPO | Cloning vector; ApR; KmR | Invitrogen |
| pDM4 | A suicide vector with ori R6K sacB and CmR | (48) |
| pTOPO::ΔVv1_2074 | pCR2.1TOPO having 2 kbs fragment comprised of each 1 kb from the upstream and downstream of complex I (Vv1_2074) | This study |
| pTOPO::ΔVv1_2074 revertant | pCR2.1TOPO having 3.29 kbs fragment of each 1 kb from the upstream and downstream with full length of complex I | This study |
| pDM4::ΔVv1_2074 | pDM4 having 2 kbs DNA fragment comprised of each 1 kb from the upstream and downstream of complex I | This study |
| pDM4::ΔVv1_2074 revertant | pDM4 having 3.29 kbs DNA fragment comprised of each 1 kb from the upstream and downstream with complex I | This study |
Quantitative real-time polymerase chain reaction (qRT-PCR)
Transcriptional levels of the complexes I to IV genes, pivotal for PMF generation, were evaluated using qRT-PCR techniques. The reference gene gyrA, was employed for qRT-PCR normalization, and primer pairs for forward and reverse sequences were designed and listed in Table 2. RNA extraction from log-phase bacterial cells cultivated in either 2.5% NaCl HI broth (in vitro) or the rat peritoneal cavity (in vivo) was performed using the RNeasy minikit (Qiagen, Germany). Subsequently, one microgram of purified RNA underwent reverse transcription into cDNA utilizing the QuantiTect Reverse Transcription Kit (Qiagen, Germany) per the manufacturer’s instructions. The qRT-PCR was executed to assess the expression levels of each target transcript employing the QuantiTect SYBR Green PCR kit (Qiagen, Germany). Relative gene expression levels were normalized to gyrA expression utilizing the threshold cycle (ΔΔCT) method.
Table 2.
Primers used in this study. C-I, Complex I; C-II, Complex II; C-III, Complex III; C-IV, Complex IV; respectively. Underlined sequences indicate the restriction enzyme recognition sites
DNA manipulation
DNA manipulation procedures followed established protocols (11) and adhered to manufacturer recommendations. Plasmid DNA purification was conducted using the QIAprep® Spin Miniprep Kit (Qiagen, Germany), while DNA fragments were purified from agarose gels using the QIAquick® Gel Extraction kit (Qiagen, Germany). Sequences were queried against the GenBank database and KEGG using the BLAST algorithm (NCBI). DNA fragment sequencing was outsourced to Macrogen Online Sequencing Order System (http://dna.macrogen.com/kor/) (Macrogen, South Korea).
Construction of NADH dehydrogenase (ΔVv1_2074) deletion mutant
To validate our hypothesis regarding the role of NADH dehydrogenase in energizing ATP synthase I for energy production, we generated a deletion mutant of the Vv1_2047 gene (Fig. 1A) using the allelic exchange method (12). PCR reactions were conducted using primers listed in Table 2, designed with overhangs recognized by specific restriction enzymes (REs) for subsequent ligation into appropriate vectors. Upstream and downstream 1000 base pair (bp) fragments of the target gene were individually amplified and then fused into 2 kb fragments through cross over PCR (13). The resulting fusion fragments were cloned into the TOPO® TA vector (TOPO® TA Cloning® Kit, Invitrogen, UK) as listed in Table 1, and positive colonies were selected via blue/white screening. Following overnight amplification in E. coli DH5α, plasmid purification was carried out, followed by digestion with appropriate REs. The resulting DNA fragments from RE digestion were then purified and subcloned into the pDM4 suicide vector (Table 1), which was subsequently transformed into E. coli SY327 λ pir. Confirmation of successful subcloning was achieved through PCR. The resulting recombinant vector was then transferred into E. coli SM10 λ pir and subsequently introduced into V. vulnificus CMCP6 via conjugation. Stable CmR transconjugants were selected on TCBS agar plates supplemented with Cm. Transconjugants were then plated on 2.5% NaCl-HI agar plates containing 10% sucrose to select clones that underwent the second homologous recombination event, leading to excision of the vector sequence and retention of only the mutated or wild-type allele of the gene. Each in-frame deletion mutation was confirmed by PCR using chromosomal DNA from the respective mutant or wild-type strain as a template (Fig. 2) and as listed in Table 1.
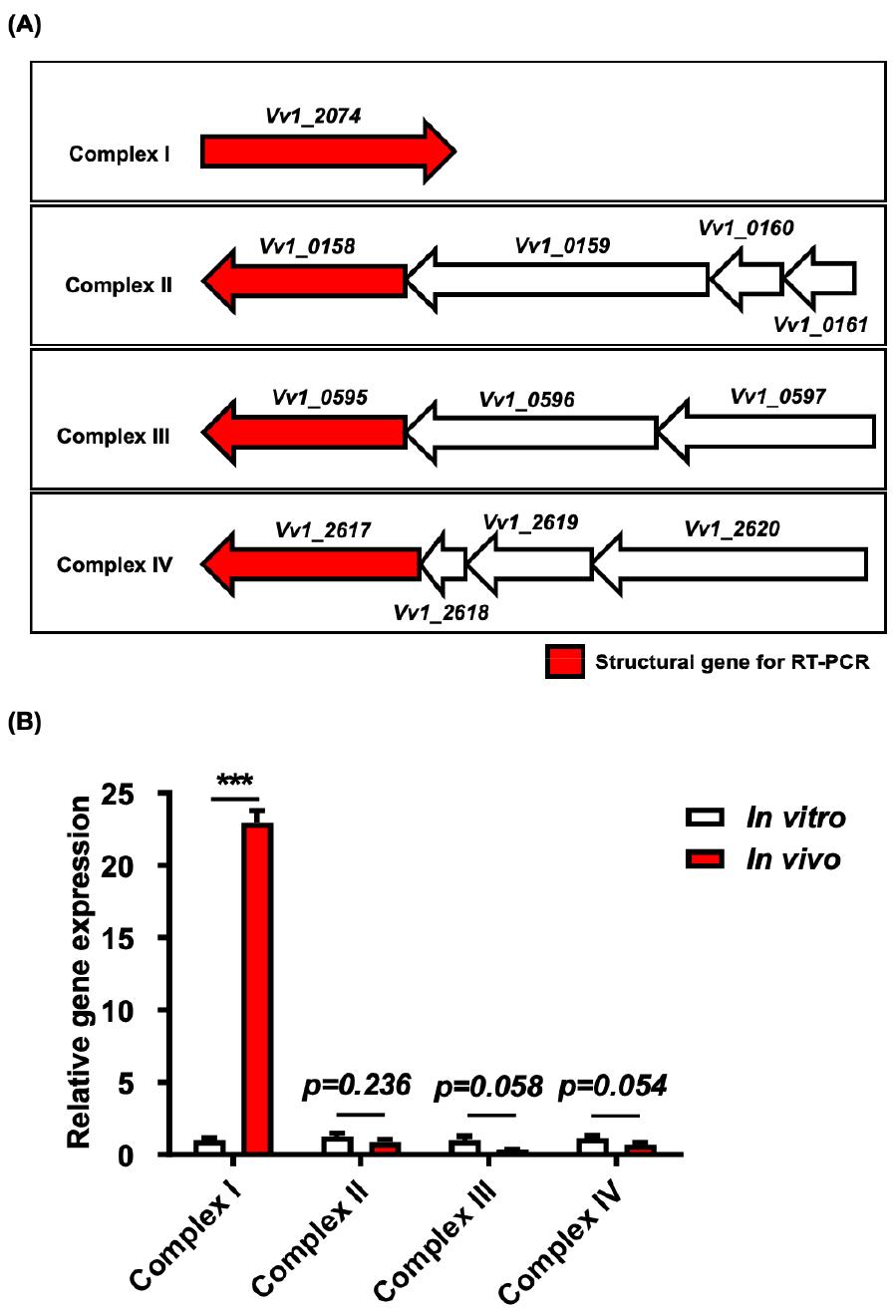
Fig. 1
Transcriptional analysis of structural genes associated with complexes I to IV in V. vulnificus CMCP6. (A) The gene and gene clusters of complexes I to IV, with red-filled genes indicating the target structural genes for qRT-PCR. Total RNA was extracted from bacteria cultured in either the rat peritoneal cavity (in vivo) or 2.5% NaCl HI broth (in vitro), followed by conversion to cDNA. The qRT-PCR was then conducted, with data normalization performed relative to the expression of the gyrA gene. (B) The relative transcript abundance values, expressed as mean-normalized gene expression relative to the in vitro expression values. Error bars denote the standard errors of means from three independent experiments. Statistical analysis was conducted using Student’s t-test (***, P < 0.001).
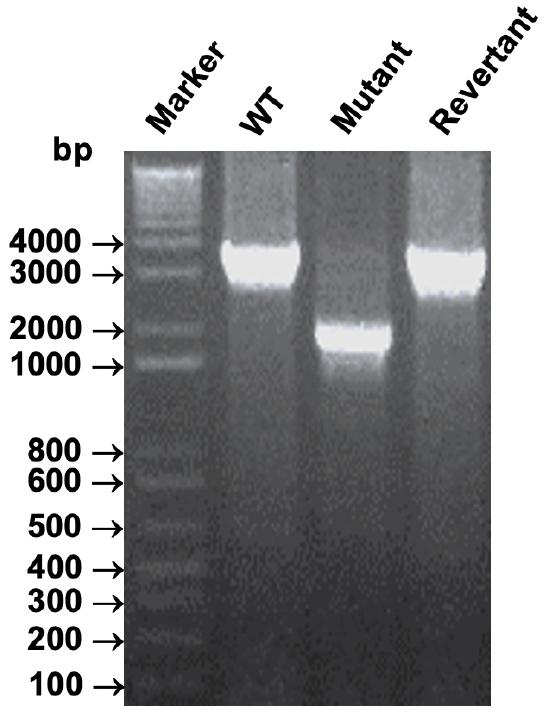
Fig. 2
The confirmation of the NADH dehydrogenase (ΔVv1_2074) deletion mutant and its revertant strains. Each in-frame deletion mutation or its corresponding revertant strain was validated via PCR analysis, utilizing chromosomal DNA extracted from the ΔVv1_2074 mutant (Mutant), ΔVv1_2074 revertant strain (Revertant), or wild-type (WT) strain as a template.
Construction of NADH dehydrogenase deletion (ΔVv1_2074) revertant strains
To verify the in vivo functions of the Vv1_2074 gene, we undertook the construction of a revertant strain of Vv1_2074, adhering to the principles of molecular Koch’s postulates (12). For precise PCR amplification, we utilized KOD Plus NEO polymerase, renowned for its proofreading capabilities (TOYOBO Life Science, USA), to amplify the entire open reading frame (ORF) of Vv1_2074. This involved amplifying regions spanning 1000 base pairs upstream to 1000 base pairs downstream using the primer pairs Vv1_2074 (Up) and Vv1_2074 (Down) as listed in Table 2. Subsequently, the amplified DNA fragments with a total 3290 base pairs were inserted into the TOPO® TA vector (TOPO® TA Cloning® Kit, Invitrogen, UK) (Table 1), and positive colonies were identified through blue/white screening. After overnight amplification in E. coli DH5α, the plasmids were purified and digested with appropriate restriction enzymes (REs). The resulting DNA fragments were then purified and inserted into the pDM4 suicide vector (Table 1). Following this, sequencing was performed using the Macrogen Online Sequencing Order System (http://dna.macrogen.com/kor/) (Macrogen, South Korea), and the obtained sequences were compared to wild-type sequences using Bio Edit 7.2 software. Upon confirmation of the DNA fragment containing the entire Vv1_2074 ORF with 1000 base pairs upstream and 1000 base pairs downstream, we proceeded with the homologous recombination procedure. This involved introducing pDM4 containing the wild type ORF of Vv1_2074 with 1000 base pairs upstream and 1000 base pairs downstream (3290 bps in total) into E. coli SM10 λ pir, followed by transfer into the ΔVV1_2074 mutant via conjugation. The resulting revertant strain was validated through PCR (Fig. 2) and as listed in Table 1.
Growth measurement of bacteria under oxygen limited culture conditions
To evaluate bacterial growth under oxygen-limited conditions, overnight cultures of V. vulnificus in 2.5% NaCl HI broth were subcultured for 4 hours at 37°C with agitation (200 rpm). Subsequently, the bacteria were washed twice with phosphate-buffered saline (PBS, pH 7.2) and inoculated into fresh 2.5% NaCl HI broth at a final concentration of 105 CFU/ml. Oxygen was depleted from the media by gas pumping. For aerobic cultures, V. vulnificus strains were inoculated into 30 mL of 2.5% NaCl HI broth and incubated at 37°C with shaking (200 rpm). Conversely, for microaerobic and anaerobic cultures, bacteria were inoculated into 6-well plates at a final concentration of 105 CFU/mL, with a final volume of 3 ml, utilizing the Campy Gas Generating Pouch System (Becton, Dickinson and Company, Sparks, USA) and the Anaerobe Gas Generating Pouch System with an indicator (Becton, Dickinson and Company, Sparks, USA), respectively (14, 15). The optical density at 600 nm (OD600) was measured spectrophotometrically at appropriate time points.
Growth of bacteria under in vivo culture conditions
The in vivo growth of the NADH dehydrogenase (ΔVV1_2074) deletion mutant and its revertant strain was evaluated relative to their isogenic wild type strain using the dialysis tube implantation assay (5). Prior to implantation, dialysis tubes (CelluSep® H1, Membrane Filtration Products, Seguin, TX, USA) were activated with phosphate-buffered saline (PBS) overnight. The dialysis membrane was then disinfected with 70% alcohol for 1 hour and washed three times with sterile PBS. Eight-week-old female Sprague Dawley rats were anesthetized using a mixture of 10% Zoletil (Virbac, Carros, France) and 5% Rompun (Bayer Korea, South Korea) dissolved in PBS. Each 5 cm dialysis tube, containing 2 mL of bacterial cells at a concentration of 5 x 105 CFU/mL, was surgically implanted into the rat peritoneal cavity. Bacterial cells were harvested at 4 hours post-implantation for viable cell counting on 2.5% NaCl HI agar plates. The growth of bacteria at 4 hours post-implantation was compared to bacterial growth under in vitro growth conditions as controls.
Determination of intracellular ATP levels
To investigate the role of NADH dehydrogenase in energy production, intracellular ATP levels of the (ΔVV1_2074) mutant strain were directly quantified using the ATP Determination kit (Invitrogen, Eugene, OR, USA) in accordance with the company’s recommendation. ATP concentration was assessed employing recombinant firefly luciferase and its substrate D-luciferin, and measured using a luminometer (MicroLumat Plus LB 962, Berthold, Germany) (16, 17, 18, 19). Cells in the mid-logarithmic growth phase at 4 hours from each culture condition (in vitro or in vivo) were used for the assay. Ten milliliters of bacterial culture from each growth condition were harvested by centrifugation at 13,000 rpm for 10 minutes at 4°C and washed once with 0.9% NaCl saline buffer (NSB). The resulting cell pellet was suspended in 500 µl of NSB containing 0.1% SDS and briefly sonicated on ice. The sonicated cell lysate was then utilized to determine the intracellular ATP concentration. The total volume of the assay was maintained consistent with the ATP standard curve assays, ensuring that the amount of sample added to the reaction mixture remained less than 10% of the total assay volume. The intracellular ATP concentration was calculated from the standard curve generated using a control ATP sample and normalized by cell number on plates. Data analysis and graph plotting were performed using Excel 2010 and GraphPad Prism 8.00 software.
Statistical analyses
The results are expressed as the mean ± standard error of the mean (SEM) values unless otherwise stated. Student’s t-test was used to compare two groups. Statistical analyses were performed using GraphPad Prism 8.00 software for Windows (GraphPad software, San Diego, CA). P values < 0.05 were considered statistically significant.
RESULTS
Quantification of the transcriptional levels of complexes I to IV in V. vulnificus infecting rat peritoneal cavity by qRT-PCR
To validate our previous systems biology results, we first examined the in vivo transcriptional profiles of structural genes related to complexes I to IV (Fig. 1A) using a rat peritoneal infection model. The qRT-PCR analysis revealed a substantial increase in NADH dehydrogenase (Vv1_2074) mRNA levels, indicative of complex I, when V. vulnificus was cultured in vivo, with a corresponding approximately 22-fold increase (P < 0.0007) (Fig. 1B). Conversely, transcript levels of complexes II (Vv1_0158), III (Vv1_0595), and IV (Vv1_2617) exhibited a decrease during in vivo growth (Fig. 1B) (P = 0.2368 for complex II, P = 0.0579 for complex III, and P = 0.0538 for complex IV). This led us to hypothesize that the highly up-regulated NADH dehydrogenase (Vv1_2074) may contribute to the PMF required by ATP synthase I. To further validate this hypothesis, we carried out a molecular genetic characterization of this gene.
Generation of NADH dehydrogenase (ΔVv1_2074) deletion mutant and its revertant strains
We generated a deletion mutant of NADH dehydrogenase (Vv1_2074) and its corresponding revertant strain, adhering to the principles of molecular Koch’s postulates (12). PCR analysis using chromosomal DNA from the mutant and revertant strains confirmed the in-frame deletions and reversions. Notably, PCR products of approximately 3 kb were observed for the wild type and revertant strains, whereas only a 2 kb PCR product was detected for ΔVv1_2074 (Fig. 2). These results affirm the successful construction of both the deletion mutant and its revertant strain.
Growth measurement of bacteria under oxygen limited culture conditions
Complex I, the primary respiratory complex, plays a crucial role in generating the PMF, essential for ATP synthesis (20, 21), across diverse species (8, 22, 23, 24). In oxidative phosphorylation, complex I enables energy production by oxidizing NADH generated from glucose glycolysis in the cytoplasm, producing NAD+ and H+(20). Subsequently, these protons are transported across a membrane, establishing a proton gradient in the extracellular membrane, facilitating electron transfer to acceptors like oxygen (9, 23, 25, 26). Hence, the absence of electron acceptors would hinder V. vulnificus from generating intracellular ATP. Therefore, we compared the in vitro growth of bacteria under various culture conditions: aerobic, microaerobic, and anaerobic conditions. Over the 24-hour period of OD600 measurement, the growth of the NADH dehydrogenase (ΔVv1_2074) deletion mutant in vitro was indistinguishable from its isogenic wild type (data not shown). Overall, it appears that NADH dehydrogenase does not play any significant role in growth under in vitro conditions.
In vivo growth retardation of NADH dehydrogenase (ΔVv1_2074) deletion mutant
Given the notable up-regulation of NADH dehydrogenase expression under in vivo conditions, we hypothesized its active involvement in infection processes. To evaluate this, we conducted in vivo growth assays using a rat peritoneal infection model. Each rat was implanted with a dialysis tube containing 2ml of 5 X 105 CFU/mL of bacterial strains. After 4 hours of incubation, implanted bacteria were harvested and viable cell counts were performed on 2.5% NaCl HI plates, with in vitro growth condition serving as the control. Data represent averages from triplicate experiments. Significantly, we observed a pronounced in vivo growth retardation of the mutant strain (P < 0.0006), which was fully restored upon reversion of the mutated gene and exhibited no defects under in vitro cultures (Fig. 3). These findings suggest the essential role of V. vulnificus NADH dehydrogenase in cell survival under in vivo conditions.
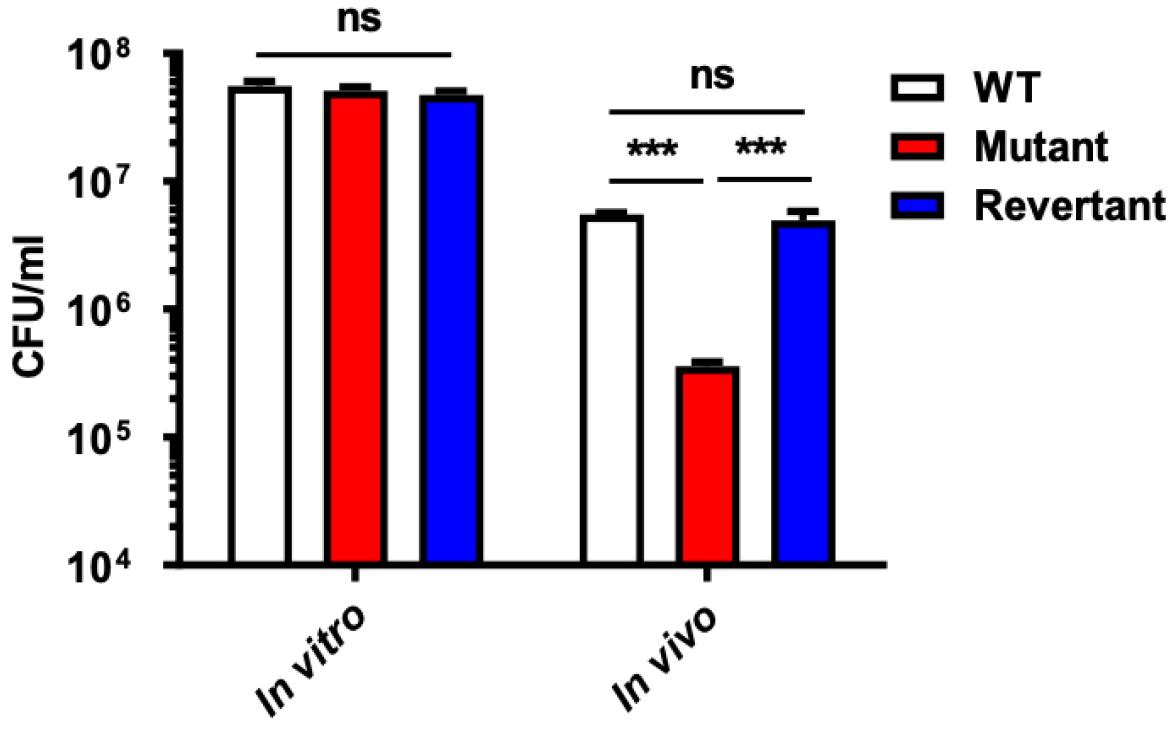
Fig. 3
Growth retardation of deletion mutant in vivo. The in vivo growth of the NADH dehydrogenase (ΔVV1_2074) deletion mutant (Mutant) and its revertant strain (Revertant) was assessed relative to their isogenic wild-type strain (WT) using the dialysis tube implantation assay. Each 5 cm dialysis tube, containing 2 mL of bacterial cells at a concentration of 5 x 105 CFU/mL, was surgically implanted into the rat peritoneal cavity. Bacterial cells were harvested at 4 hours post-implantation for viable cell counting on 2.5% NaCl HI agar plates. The growth of bacteria at 4 hours post-implantation was compared to bacterial growth under in vitro growth conditions as controls. The error bars represent the standard errors of means from three independent experiments. Statistical analysis was carried out using Student’s t-test (***, P < 0.001; ns, non-statistically significant difference).
Significant reduction of intracellular ATP in NADH dehydrogenase (ΔVv1_2074) deletion mutant under in vivo conditions
To elucidate the role of NADH dehydrogenase in ATP production by V. vulnificus, we directly measured intracellular ATP levels in the mutant strain across various growth conditions. Cells in mid-logarithmic growth phase from each culture condition (in vitro or in vivo) were utilized for the assay. Intracellular ATP concentration was determined from a standard curve generated using control ATP samples and adjusted by viable cell number for comparison. Data represent averages from triplicate experiments. Remarkably, we observed no significant reduction in intracellular ATP levels in the mutant strain under in vitro growth condition. Nevertheless, under in vivo growth conditions, a notable decrease in intracellular ATP levels was observed (P < 0.0001) (Fig. 4), which was fully reinstated in the revertant strain. These findings strongly indicate that NADH dehydrogenase contributes to energy production specifically during established V. vulnificus infection in vivo.
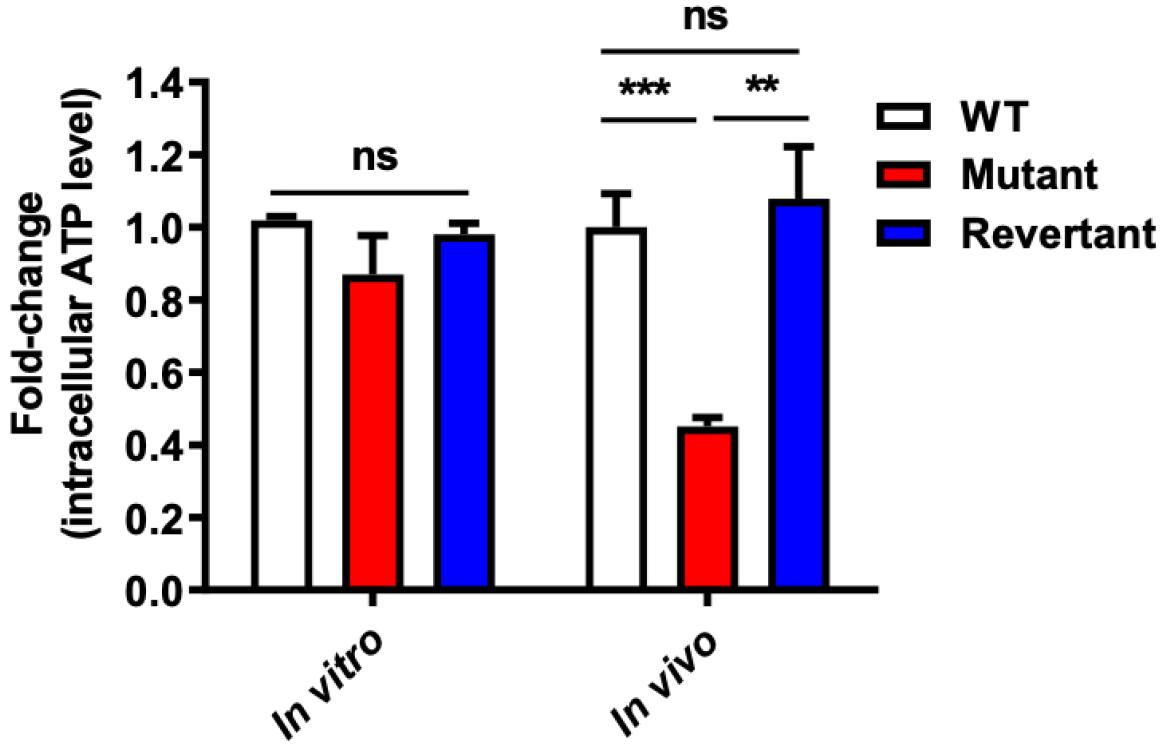
Fig. 4
Significant reduction of intracellular ATP in deletion mutant from in vivo growth conditions. The intracellular ATP concentration of the NADH dehydrogenase (ΔVV1_2074) deletion mutant (Mutant) and its revertant strain (Revertant) was quantified relative to their isogenic wild-type strain (WT) using recombinant firefly luciferase and D-luciferin substrate. Cells in the mid-logarithmic growth phase at 4 hours from each culture condition (in vitro or in vivo) were utilized for the assay. Intracellular ATP concentration was determined from the standard curve generated using a control ATP sample and normalized by cell number on plates. Data analysis and graph plotting were calculated using Excel 2010 and GraphPad Prism 8.00 software. The error bars represent the standard errors of means from four independent experiments. Statistical analysis was performed using Student’s t-test (**, P < 0.01; ***, P < 0.001; ns, non-statistically significant difference).
DISCUSSION
The shift in V. vulnificus’ environment during infection, transitioning from its natural estuarine habitat to a host milieu (1, 27, 28, 29), prompted us to investigate potential disparities in ATP generation mechanisms. Leveraging our previous in vivo transcriptome and proteome analyses conducted using a rat peritoneal infection model, we uncovered significant alterations in gene expression and flux patterns (4, 5). Specifically, we observed a marked up-regulation of the NADH dehydrogenase (complex I) gene, alongside a concurrent down-regulation of succinate dehydrogenase (complex II), cytochrome bc (complex III), and cytochrome oxidase (complex IV) in vivo. These observations stood in contrast to the established roles of the five membrane-bound complexes in electron transport and oxidative phosphorylation for energy production (6, 7, 8, 9). Moreover, our analyses revealed increased flux analysis of ATP synthase I, highlighting its pivotal role in energy production post-infection. Together, these findings suggest distinct ATP generation mechanisms for V. vulnificus in vivo compared with in vitro cultures, prompting our hypothesis regarding the potential involvement of NADH dehydrogenase (complex I) as a proton source for ATP synthase I post-infection.
Construction of a deletion mutant of NADH dehydrogenase (ΔVv1_2074), along with its revertant strain (Fig. 2), enabled us to assess its contribution to energy production. Despite observing no significant differences in growth or intracellular ATP levels of the mutant strain under in vitro conditions, a significant growth retardation and reduction in intracellular ATP were evident under in vivo conditions, both of which were reversed upon reversion of the mutated gene (Figs. 3-4), respectively. These results suggest the essentiality of complex I of V. vulnificus for cell survival in vivo, suggesting its pivotal role in contributing PMF to energy production via complex V.
Previous studies on the oxidation and phosphorylation pathways in prokaryotic and eukaryotic cells have consistently demonstrated the ubiquitous presence of NADH dehydrogenase across all subdivisions of proteobacteria and closely related forms of this complex have been identified in the mitochondria of eukaryotes and in the plasma membranes of purple bacteria (10, 21, 22, 25, 30). NADH dehydrogenase plays a crucial role in generating the PMF, which is essential for ATP synthesis (8, 31, 32, 33, 34, 35, 36). Interestingly, most of bacteria derive energy through the transfer of electrons from a low-potential substrate, such as glucose, to a high-potential electron acceptor, such as molecular O2. This process is commonly known as respiration (6, 23, 25, 26, 37) and the coupling of electron transport in oxidative phosphorylation for ATP generation via ATP synthase I has also been elucidated in human mitochondria (38, 39). Noticeably, it is well-known that the PMF directly supplies energy to ATP synthase I for the generation of intracellular ATP (8, 32, 33, 34, 35, 36). Those studies strongly corroborated our findings, indicating that the deletion mutant of complex I in V. vulnificus CMCP6 exhibited a significant reduction in intracellular ATP levels under in vivo conditions. Furthermore, our results highlight the essential role of complex V as an energy source under O2 limited conditions (unpublished data).
It is widely understood that under aerobic conditions, not only complex I but also other complexes (II, III, and IV) are responsible for electron transport and proton pumping from the cytoplasmic side to the extracellular space, as demonstrated in studies on yeast (24) and E. coli(40) and complex V is responsible for the inward movement of protons into the cell, facilitating ATP synthesis (41, 42, 43, 44, 45, 46). However, our in vivo transcriptome analysis revealed that only complex I was up-regulated in vivo, while complexes II, III, and IV appeared to be down-regulated after infection. To confirm the gene expression levels of complexes (I-IV) in vivo, we conducted qRT-PCR analysis. The results showed that among the PMF-related genes, complex I exhibited significant up-regulation in vivo, while complexes II-IV displayed down-regulation under in vivo culture conditions (Fig. 1B), consistent with our transcriptome analysis. This differential gene expression pattern provided additional evidence that complex I plays a dominant role in PMF generation in vivo, a finding supported by other studies (8, 31, 32, 33, 34, 35, 36). Additionally, the intracellular ATP levels of the NADH dehydrogenase deletion mutant under in vitro conditions were not significantly reduced. This suggests that other complexes (II-IV) may contribute to ATP synthesis by providing a charge to complex V, although they were not preferentially expressed under in vivo conditions. However, future experiments will focus on further elucidating the mechanisms of NADH dehydrogenase in energy metabolism and its interaction with other complexes, including comprehensive in vivo analyses to validate these findings.
Our study sheds light on the intricate interplay between NADH dehydrogenase and ATP synthase I in V. vulnificus energy metabolism post-infection. Despite the need for further elucidation of the precise mechanisms governing PMF generation by NADH dehydrogenase or other complexes (II-IV) under in vitro and in vivo, our findings provide compelling evidence for NADH dehydrogenase’s significance in energy production during infection. In conclusion, the essential role of NADH dehydrogenase (complex I) in ATP synthesis under in vivo conditions highlights its potential as a therapeutic target for combating V. vulnificus infections. Furthermore, our study contributes to the growing body of evidence supporting the phenomenon of metabolic reprogramming post-infection and bolsters the validity of our systems biological analyses.
