

INTRODUCTION
Varicella-zoster virus (VZV) belongs to the subfamily Alphaherpesvirinae of the Herpesviridae family, and is the smallest and genetically stable among eight human herpes viruses. VZV has a genome of about 125 Kbp, linear double-stranded DNA, and encodes 74 open reading frames (ORFs). The first VZV strain whose entire nucleotide sequence was identified is Dumas (NC001348), which is used as a reference in research and analysis of VZV (1).
VZV causes varicella as a primary infection and exists as a latent infection in sensory ganglia. Afterwards, if the immune system is weakened or in the elderly, the virus reactivates and causes herpes zoster (2, 3). Aerosolized varicella virion from the lesion enters the upper respiratory tract via droplets and infects T cells (4). Infected T cells migrate to the skin via blood, multiply and cause rashes and blisters, which is called chickenpox. Infectivity is maintained for 3-7 days after the rash appears or until the blisters become crusted. Afterwards, when the cellular immunity weakens the virus dormant in the sensory ganglion reactivates and forms a band-shaped herpes on one side of the face or body, which is called herpes zoster. Rash caused by herpes zoster usually disappears within 2-4 weeks, after which the neuralgia may persist for months to years, which is called postherpetic neuralgia (5, 6). Chickenpox occurs predominantly in children and occurs in 50% of children before the age of 5, and is experienced in 90% by the time they are 12 years of age (7). In addition, it is highly contagious, and is often transmitted through family contact. In Korea, chickenpox was designated as a legal communicable disease in 2005, and the chickenpox vaccine was included in the basic vaccination.
Varicella and zoster are currently prevented by live attenuated vaccines. A wild strain was isolated from a 3-year-old boy who had chickenpox in Japan in 1974, and attenuated through subculture to develop a vaccine called vOka (8). The varicella vaccine was commercialized in Japan from 1988. Afterwards, modified formulation based on vOka was developed by Merck and GlaxoSmithKline in the United States (9, 10), and basic inoculation was performed from 1995 (11). In Korea, Green Cross has developed a chickenpox vaccine called SuduVax using the virus isolated from a 33-month-old boy with chickenpox in Seoul (12). SuduVax was sold in Korea in 1994 and commercialized internationally in 1998.
Attenuated varicella vaccine produced through subculture is used throughout the world, but the mechanism of vaccine attenuation has not been precisely elucidated. Characteristics of the attenuated vaccine development process include non-natural culture conditions such as low temperature culture and culturing of the virus in guinea pig cells. In this study, we tried to understand the attenuation mechanism by comparing clinical strains subcultured in non-natural culture conditions at low temperature and in guinea pig cells with those cultured in natural conditions.
MATERIALS AND METHODS
Virus culture
In a previous study, YC02, a clinical strain of VZV, was isolated from a 3-year-old boy who developed chickenpox (13). YC02 was grown in human foreskin fibroblast (HFF, passage 16-22) cells at 37°C and 5% CO2. In order to replicate at low temperature, YC02 at passage 17 (p17) was subcultured in HFF cells at 34°C until passage 60. For replication of YC02 in guinea pig cells, YC02 cultured at 34°C until passage 30 (p30 (34)) was transferred to 37°C. After 4 passage at 37°C, virus was inoculated into guinea pig lung cells (GPLC, ATCC No. CCL-158, passage 34-40) and cultured at 37°C. Serial passaging scheme of VZV YC02 in different culture conditions is summarized in Fig. 1.
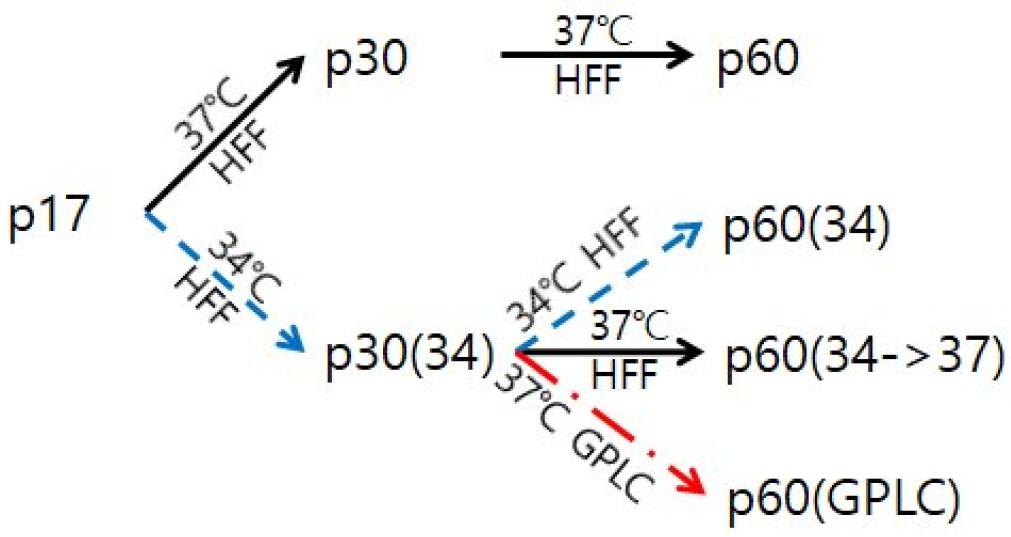
Fig. 1
Overview of serial passaging of VZV. HFF: human foreskin fibroblast; GPLC: guinea pig lung cell.
Dulbecco’s modified Eagle medium (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and F-12 nutrient mixture medium (Gibco) containing 10% fetal bovine serum (FBS, Gibco), 100 μg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA) and 100 U/ml penicillin (Sigma-Aldrich) were used for HFF and GPLC, respectively. Cells grown as a monolayer in a cell culture flask was inoculated by the virus diluted in a ratio of 1:8. After 1 hr adsorption at 37°C in 5% CO2, virus inoculum was removed and a medium containing 2% FBS was added. The virus was subcultured until passage 60.
Virus harvest and concentration
Virus samples were harvested at every 5 passages for infectivity assay or whole genome sequencing. When the virus-infected cells showed a cytopathic effect (CPE) of 80% or more, the medium was removed, washed once with phosphate-buffered saline (PBS, pH 7.4), and trypsin-ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich) was added. After incubation at 37°C for 3 min, the cells were suspended in serum-free DMEM, and centrifuged at 800 xg for 10 min. The cell pellet was suspended in a mixture of 90% cell-freezer (Genenmed, Seoul, Korea) and 10% dimethylsulfoxide (Sigma-Aldrich), and stored in liquid nitrogen until further assay. Even if CPE did not appear in the virus-infected GPLC, the virus was harvested in the same way 3-4 days after virus inoculation, and F-12 medium was used instead of DMEM.
Since virus samples obtained from infected cells contain large amounts of cellular debris in addition to the viral genome, it was necessary to remove cellular impurities as much as possible and to concentrate the virus for genome sequencing. For this, the freezing & thawing process was repeated 3 times followed by 12 cycles of sonication (Sonics, VCX130, Newton, CT, USA) at an amplitude of 20% for 10 seconds. The sonicated samples were centrifuged at 8,000 rpm for 5 min at 4°C. For virus concentration, 0.8 g/ml ammonium sulfate ((NH4)2SO4) was mixed 1:1 with the supernatant, stirred for 4 hrs, and centrifuged at 10,000 xg, 4°C for 30 min. The precipitate was suspended in 200 μl of serum-free medium.
DNA extraction and next generation sequencing (NGS)
DNA was extracted from the concentrated virus using Exgene™ Cell SV mini kit (GeneAll Biotechnology, Seoul, Korea). To estimate the amount of viral DNA and cellular DNA, q-PCR was performed with IQ SYBR Green SuperMix (BIO-RAD, Hercules, CA, USA). Using previously known references, two pairs of primers capable of detecting cellular DNA and VZV DNA were used. To check the amount of cellular DNA, a primer that amplifies the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) site was used, and to check the amount of viral DNA in VZV, a primer that amplifies the ORF4 site was used (14, 15). PCR was repeated 45 times under the conditions of initial denaturation at 94°C for 4 min, denaturation at 94°C for 30 seconds, annealing at 48.9°C for 30 seconds, and extension at 72°C for 30 seconds.
Whole genome sequence of the extracted viral DNA was determined with Illumina’s Hi-Seq 2500 NGS system (San Diego, CA, USA), and this process was carried out by Theragen Etex (Suwon, Korea). Illumina TruSeq DNA Sample Preparation kit (v2) was used to prepare the DNA library, and the prepared library was sequenced in the Hi-Seq method. After quality check and data trimming with Illumina version 1.8, we obtained mapped reads between 244,242 and 3,449,279 and read depth between 196 and 2,768. For reads with guaranteed quality, mapping assembly was performed using the clinical strain YC02 (MF004348) as a reference sequence. As a result of mapping assemble, one complete nucleotide sequence was obtained.
Genomic analysis
Information on open reading frame (ORF) and non-coding region (NCR) of each YC02 variant was explored using local blast search against VZV reference strain Dumas (NC001348). And the ORF check function of CLC Sequence Viewer (version 6.4, http://www.clcbio.com), and ORF Finder provided by NCBI (http://www.ncbi.nlm.nih.gov/) programs were utilized to check whether the searched ORF location was correct and whether there was an additional ORF location. BioEdit Sequence Alignment Editor (version 7.0.9.0, http://www.mbio.ncsu.edu/bioedit) was used to check and confirm whether the ORF has an intact start codon and stop codon in the actual nucleotide sequence.
For mutation and genetic polymorphism analysis, it was necessary to align the YC02 base sequence with respect to the standard virus strain. The virus strain used as a standard was Dumas (NC001348), the first to determine the entire nucleotide sequence among the VZV, and the entire nucleotide sequence was used. The entire base sequence and amino acid sequence for each passage were read with BioEdit and aligned using Clustal W2 (version 2.1).
Genetic polymorphism analysis
Genetic polymorphism refers to having two or more bases together at any same nucleotide position in a genome. Information on the number of A, C, G, and T bases for each position of the nucleotide sequence was obtained from the NGS analysis of the YC02 variants. Among the bases mapped for each position, the base that occupies the largest proportion is defined as the major base, and constitutes the representative or consensus nucleotide sequence. The base that occupies the second largest proportion is determined as a minor base. The Genetic Polymorphism Analyzer (GPA) program (https://github.com/lis123kr/GPA) developed in our laboratory was used for analysis. This program compares the ratio of minor bases between sequences, finds genetic polymorphisms, and shows the results of analysis in various forms. In this study only the genetically polymorphic site (GPS) having a minor base ratio of 5% or more and a read-depth of 35 or more was analyzed (16).
Virus infectivity and growth kinetics
In order to measure the infectivity of the virus, plaque assay was performed every 5 passages during subculture. The harvested virus sample (see above) was serially 10-fold diluted in serum-free DMEM. In a 6-well plate in which HFF was grown as a monolayer, 200 μl per well of the virus dilution was inoculated and adsorbed for 1 hr with gentle shaking every 15 min. Thereafter, DMEM containing 2% FBS was added and incubated. After 7 days, the medium was removed, washed once with PBS (pH 7.4), 10% formalin (in 0.85% saline) was added, and the cells were fixed overnight. The next day, after removing the formalin and staining the fixed cells using crystal violet (0.03% w/v in 10% ethanol, Sigma-Aldrich), the plaques were counted under a dissecting microscope with a 10X magnification.
Growth kinetics of YC02 variants was investigated by infecting HFF grown in 60 mm dishes with virus samples at multiplicity of infection of 0.02 plaque forming units (pfu)/cell. After one hr adsorption at 37°C, virus inoculum was removed and the infected cultures were fed with DMEM containing 2% FBS. At 1, 2, 3, 4 and 5 days post infection (d p.i.), infected cells were harvested by trypsinization and centrifuged at 800 xg. Infected cell pellets were resuspended with freezing media and stored in liquid nitrogen until plaque assay.
RESULTS
Mutations that occurred during propagation at low temperature
In order to find out what genetic variations occurred when the virus was propagated in non-natural conditions, the VZV clinical isolate YC02 was subcultured at low temperature (34°C) and non-human cells (GPLC) (Fig. 1). At the 30th passage (p30) and 60th passage (p60), the entire genome sequence of each variant was determined and compared with that of p17 before propagation in non-natural conditions. A case in which sequences with different bases are found in one or more variants is called single nucleotide polymorphism (SNP), and in this study, it was found in a total of 41 nucleotide positions (Table 1).
Table 1.
Mutations found in VZV strain YC02 subcultured in non-natural conditions
To find out what mutations occurred in the low-temperature culture, the nucleotide sequence of YC02 p17 before low-temperature culture was compared with those of p30 (34) and p60 (34) that were harvested at p30 and p60 after starting to be cultured at 34°C from p18. In p30 (34), mutations were observed in two positions, 87656 and 91239, and were not observed in any other culture conditions, so it can be said that the mutation occurred specifically during the process of the low-temperature culture (Table 1, green background). Both of these are T to C substitution mutations. Since they are T in p60 (34), it seems that a back mutation occurred during subculture of p30 (34) to p60 (34) at 34°C.
In p60 (34), mutations were observed in 37 nucleotide positions (Table 1, red letter). Among them, 26 mutations were considered to be related to low-temperature culture since mutation was not observed in p60 cultured in natural conditions. They can be further divided into 4 types (Fig. 2). In the first type, mutation was observed only in p60 (34) (Fig. 2A), and were found in 6 nucleotide positions (Table 1, yellow background). It was found in one site in ORF14 and two sites in ORF36. Two mutations in ORF36 occurred at the first and third positions of the same codon, resulting in I214V nonsynonymous substitution. The other three were found in NCR. These first types of mutations can be considered low temperature-specific mutations, occurred during propagation of p30 (34) to p60 (34) at 34°C. In the second type, same mutation was also observed in p60 (34->37) (Fig. 2B), which was found in 17 sites. In the third type, two mutations in nucleotide positions 1337 and 63539 were observed in all three p60 variants subcultured from p30 (34) (Fig. 2C). The fourth type is observed only in p60 (34) and p60(GPLC), which are non-natural culture conditions (Fig. 2D). Since it does not appear in p60 (34->37), it is thought to be a mutation that occurred under non-natural conditions.
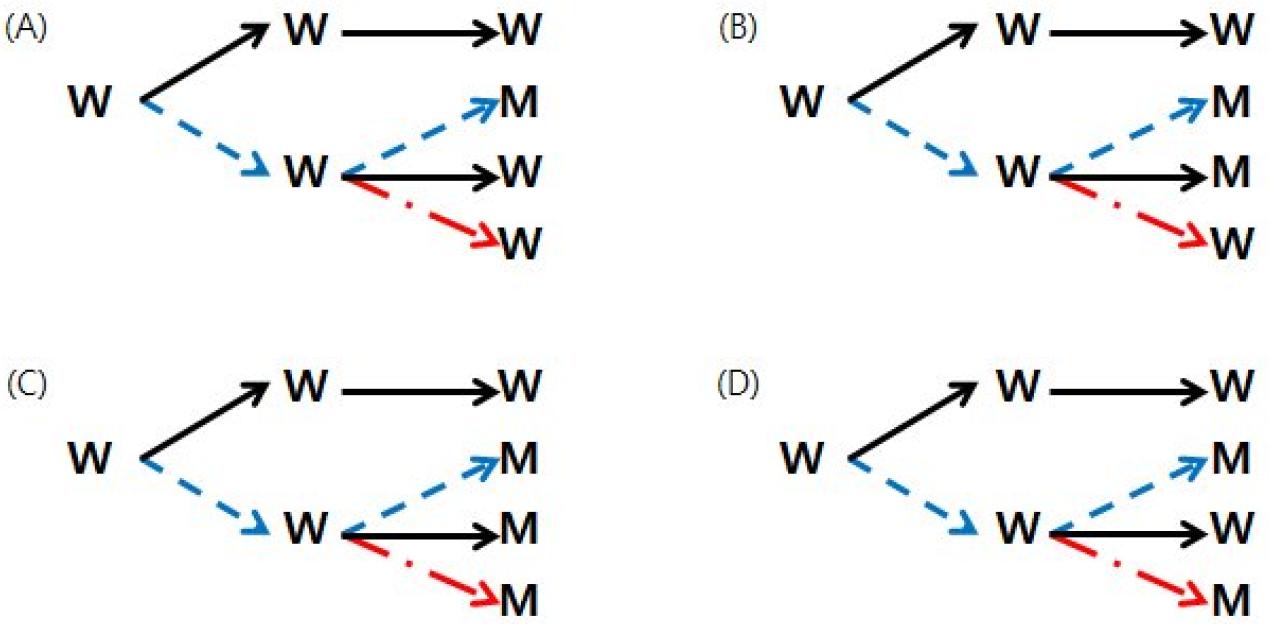
Fig. 2
Four types of mutations found in three lineages of p60 subcultures from p30 (34). (A), mutation was detected only in p60 (34), found at 6 positions: 2676, 20829, 65446, 65448, 104529, 105054. (B), mutations were detected both in p60 (34) and p60 (34->37), found at 17 positions: 2314, 4440, 9288, 13667, 20963, 41458, 41459, 66247, 80436, 88867, 97525, 100653, 105009, 106262, 107070, 107371, 107372. (C), mutations were detected in all three p60’s, found at 2 positions:1337, 63539. (D), detected in p60 (34) and p60 (GPLC), found only at 1 position: 97748.
Mutations that occurred during propagation in non-human cell culture
In p60 (GPLC), 11 nucleotide positions different from p30 (34) were observed (Table 1, green letter). There were two mutations observed only in p60 (GPLC) (Table 1, blue background). Early termination of ORF18 is expected by insertion of A at position 26470. C to T substitution at position 94135 resulted in an S617K nonsynonymous mutation in ORF54. Substitution mutations in the other 9 positions were not considered to be GPLC-specific mutations since mutations were observed in other culture conditions also.
Mutation at vaccine-specific sites
In our previous study on VZV vaccine, 24 vaccine-specific sites were suggested (13). Among them, the mutations that occurred in the non-natural conditions were found in two nucleotide positions in this study. A585T nonsynonymous mutation occurred in ORF55 due to G to A substitution at position 97748. Since the mutation in 97748 appeared both in p60 (34) and p60 (GPLC) and not in other culture conditions, it is believed that the mutation occurred during the process of subculture of p30 (34) under non-natural conditions such as low temperature (34°C) or non-human cells (Fig. 2D). T to C substitution at position 106262 causes the R958G nonsynonymous mutation in ORF62. This mutation was observed in p60 (34) and p60 (34->37) but not in p60 (GPLC). Thus, this mutation is considered to occur during the process of subculture of p30 (34) in HFF, a natural cell regardless of the culture temperature (Fig. 2B). Similar mutations were also found at 16 positions other than position 106262.
Changes in genetic diversity
Genetic diversity can be expressed in many ways, and genetic polymorphism is one of them. Genetic polymorphism refers to co-existence of two or more bases at a frequency of 5% or more at any one position on the genome. We examined how genetic polymorphism changed when VZV YC02 was propagated in non-natural conditions. The site on the genome where genetic polymorphism appears is called a genetically polymorphic site (GPS). The number of GPS increased as VZV was subcultured in natural conditions (Table 2). When subcultured at low temperature, the number of GPS decreased by about 15% compared to when subcultured at normal temperature, while the minor allele frequency (MAF) in GPS slightly increased. On the other hand, when subcultured in GPLC, the number of GPS was significantly reduced compared to when cultured in natural conditions or at low temperature (Table 2).
Table 2.
Genetic polymorphism in VZV strain YC02 subcultured in non-natural conditions
| Strain | No. GPS | Average MAF | Hs |
|---|---|---|---|
| p17 | 28 | 0.205 | 3.04 |
| p30 | 36 | 0.182 | 3.31 |
| p30 (34) | 31 | 0.201 | 3.10 |
| p60 | 110 | 0.170 | 4.48 |
| p60 (34) | 93 | 0.241 | 4.32 |
| p60 (34→37) | 101 | 0.229 | 4.38 |
| p60 (GP) | 35 | 0.170 | 3.29 |
Shannon entropy (Hs), another indicator of genetic diversity, is calculated as the sum of the MAF of each GPS divided by Ln (MAF) (17). Hs also increased with subculture in natural conditions. In the case of subculture at low temperature, the Hs was slightly lower than subculture at normal temperature (Table 2). On the other hand, in p60 (GPLC), Hs was significantly reduced. Therefore it is thought that genetic diversity is reduced when YC02 was propagated in GPLC.
Infectivity of VZV YC02 propagated in non-natural conditions
In order to examine whether the infectivity of VZV subcultured in non-natural conditions, VZV strain YC02 cultured in various conditions as shown in Fig. 1 were harvested every 5 passages from p15 to p60, and infectivity was evaluated by plaque assay. There was no significant difference in virus infectivity when cultured at low temperature compared to when cultured at normal temperature (Fig. 3). On the other hand, when cultured in GPLC, the infectivity decreased sharply, and it appeared that there was almost no infectivity between p40 and p45, and infectivity were gradually recovered thereafter.
Fig. 3
Changes in the infectious virus titer with propagation of VZV in natural and non-natural conditions in vitro. The VZV strain YC02 was propagated in HFF and GPLC culture to up to 60 passages. At every 5 passage, the infectious titer was determined by plaque assay. pfu, plaque-forming units. (-□-) HFF at 37°C, (-○-) HFF at 34°C, (-△-) HFF at 34°C and shifted to 37°C, (-◇-) GPLC at 37°C.
The proliferative ability of viruses subcultured up to passage 60 in various culture conditions was investigated using growth kinetics assay in HFF cells. The proliferation of p60 cultured in natural conditions was the highest, and the proliferation of p60 (GPLC) cultured in guinea pig cells was the lowest (Fig. 4). The proliferation of p60 (34) cultured at a low temperature is slightly lower than that of p60. The difference in proliferative ability between p60 and p60 (34) increased on the 4th and 5th days of infection. The proliferative ability of p60 (34->37) was intermediate between that of p60 and p60 (34).
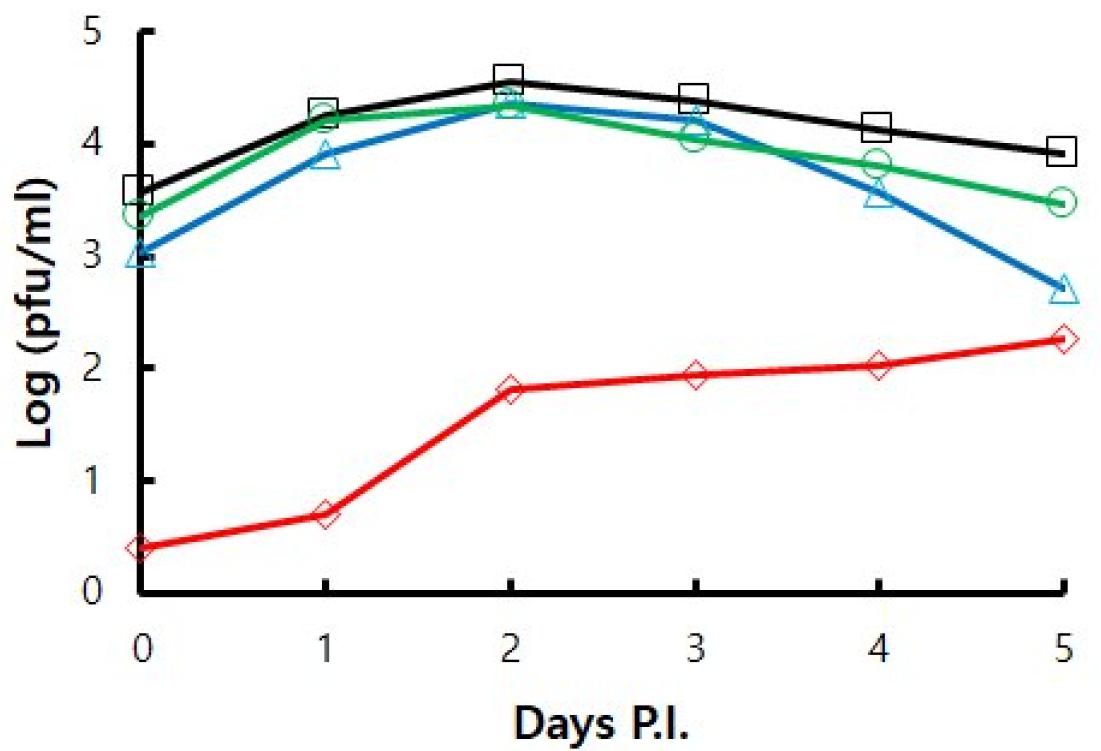
Fig. 4
Growth kinetics of VZV YC02 subcultured up to passage 60 in various culture conditions. Samples were harvested every day until 5 days post infection (Days P.I). Virus infectivity was determined by plaque assay. (-□-) HFF at 37°C, (-○-) HFF at 34°C, (-△-) HFF at 34°C and shifted to 37°C, (-◇-) GPLC at 37°C.
Discussion
Attenuation procedures for varicella vaccine production include propagation of VZV in non-natural conditions such as low temperature and non-human cell culture (8, 9, 12). Accordingly, we wanted to find out what kind of genetic and biological changes were accompanied during propagation of VZV in non-natural conditions.
One way of non-natural culture conditions is low temperature culture. Since 34°C was used in the manufacturing process of the commercial varicella vaccines, this study also tried to find out what kind of genetic change occurred when VZV was propagated at 34°C. The site where p60 (34) mutation was most observed during low temperature culture was ORF62. ORF62 is a gene encoding a VZV transactivator protein (18, 19), and is known as the main mechanism of attenuated vaccines by inhibiting the proliferation and spread of viruses when mutations occur in this region (20). However, the mutations found in ORF62 are difficult to consider as low temperature-specific because they are also found in other culture conditions including p60 cultured at normal temperature and p60 (34->37) cultured at a low temperature and shifted to normal temperature.
Low temperature-specific mutations are considered to include two sites that were observed only in p30 (34) and six sites that were observed only in p60 (34). The two sites (positions 87656, 91239) observed only in p30 (34) appeared to undergo a back mutation when continuously cultured in various conditions including normal temperature, low temperature, and non-human cells, so it is difficult to consider it a low temperature-specific mutation. On the other hand, the six mutations observed in p60 (34) did not appear in other culture conditions, thus they can be considered to be low temperature-specific mutations.
In previous studies comparing the genome sequences of vaccine and clinical strains, vaccine-specific sequences thought to be related to vaccine attenuation have been suggested (13, 21, 22). Two of them were identified in this study. Since the G to A mutation at position 97748 appeared in both p60 (34) and p60 (GPLC) but not in other culture conditions, it is thought that the mutation occurred when p30 (34) was subcultured in non-natural conditions. Attenuation of commercial varicella vaccines includes low-temperature culture and non-human cell culture (9, 12, 23). Therefore, it is thought that the mutation at position 97748 is closely related to propagation in non-natural conditions. The T to C mutation at position 106262 was observed in p60 (34) and p60 (34->37), thus it could be considered to be a low temperature-specific mutation. However, same T to C mutation at this position occurred even when YC02 and another clinical strain, YC01, were cultured up to p110 in natural culture conditions (24). Therefore, it is controversial whether the T to C mutation at position 106262 is a low temperature-specific mutation.
There was no significant difference in the infectivity and proliferation of the virus cultured at low temperature compared to the virus cultured at normal temperature. On the other hand, the infectivity and proliferative power of viruses cultured in GPLC were significantly reduced compared to viruses cultured in natural HFF cells. The infectivity of YC02 subcultured in non-natural GPLC was decreased but recovered gradually later. It seemed that a very small portion of the virus survived and adapted to GPLC culture. And the adapted virus began to propagate in GPLC. Similar observation was reported previously. Extensive subculture of VZV in HFF cells appeared to induce adaptation to non-natural GPLC culture (24). Genetic diversity showed similar results. Perhaps the low temperature of 34°C does not give much stress to virus replication compared to 37°C, but it seems to be greatly affected when the virus grows in GPLC. In addition to YC02, our laboratory has two more clinical strains, YC01 and YC03 (25). Two clinical strains other than YC02 did not proliferate in GPLC at all, so they were not included in this study.
Many mutations occurred during culture under non-natural conditions such as low temperature and non-human cells. But considering other culture conditions, it is thought that low temperature-specific mutation was found at 6 positions and GPLC-specific mutation at 2 positions. Among them, the mutation that is thought to affect the immune response is the K95N nonsynonymous mutation in ORF14. ORF14 encodes gC. VZV gC is a type 1 transmembrane protein and its ectodomain binds to chemokine to increase migration of leukocytes including tonsillar leukocytes, the main target of VZV primary infection (26). This result seems to be related to the finding that VZV gC is not essential for in vitro replication, but the replication ability of VZV isolate lacking gC is reduced in SCID mice (27). Therefore, it will be interesting to further investigate whether the K95N mutation affects the in vitro and in vivo replication of VZV as well as the immune response to VZV infection.
The most common way to attenuate viruses is to culture them in non-natural conditions such as low temperatures and non-human cells (28). Since the varicella vaccine was also produced through the culturing process under such non-natural conditions, it was expected that many of the vaccine-specific mutations could be found through this study. However, only one mutation (position 97748) was found to be associated with a non-natural condition. One of the reason may be that the culture conditions used in this study and the culture conditions when making commercial vaccines were not identical. The passage numbers 30 and 60 used in this study were selected with the intention of making them similar to the final passage number of commercial vaccines. However, the actual passage number is 25-38 for Varivax, 35-38 for Varillrix, and 64 for Suduvax, which are somewhat different from passage numbers 30 and 60 used in this study (12, 23). Another reason is thought to be that the virus used in the experiment was different. The vaccine strain that produced Suduvax, a Korean varicella vaccine, is called MAV/06, which means that it was the 6th among several clinical strains used in the experiment (12).The other clinical isolates fell short of expectation and failed to produce attenuated vaccine candidates. In this study, clinical strains other than YC02 did not propagate in GPLC at all (data not shown). Even if several clinical strains are passaged with the same protocol, it is difficult to obtain successful attenuation from all clinical strains. In the future, if more clinical strains are cultured in more diverse non-natural conditions, it will be helpful for the study of the attenuation mechanism of the VZV vaccine.
