

INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causal pathogen of human tuberculosis (TB), remains a major killer among infectious diseases caused by single bacterial pathogens (1). Mtb can be transmitted via an aerosol and infects alveolar macrophages (AMs). Mtb is then disseminated to other myeloid cells, among which macrophages and neutrophils are known to eradicate Mtb (2). However, they also serve as niches in which Mtb may persist (2). Mtb employs several strategies to evade the host protective immune responses, thereby progressing to human TB. Mtb virulence factors include several cell surface components, enzymes, and transcriptional factors (3, 4). Among them, the ESAT-6 secretion system-1 (ESX-1), also known as the type VII secretion system, which is induced by region of difference (RD) 1, plays a critical role in TB pathogenesis (5, 6). Host cell–Mtb interactions are mediated by dynamic dialogs between Mtb and host cell components (7). The protective host response against TB is complex.
To date, several host immune and non-immune components involved in protection against intracellular Mtb have been reported. Among these host protective factors/pathways, we discuss the major immune strategies that together constitute the host defense against Mtb infection. These include TB granulomas; innate and adaptive immune responses; macrophages, neutrophils, and dendritic cells (DCs); and autophagy. Although these are all essential for protection against TB, changes in the regulation thereof may be detrimental to the host. Additionally, we discuss several cytokine biomarkers in the context of Mtb infection. It is crucial to understand the intricate controls that govern the expression of protective components that enhance host defense and prevent disease progression.
Overview of Mtb infection
Mtb, a facultative intracellular pathogen, is a rod-shaped bacillus that belongs to the slow-growing mycobacteria of the Mtb complex, which includes mycobacteria other than Mtb, and M. leprae (the leprosy pathogen) (8, 9). Although systemic Mtb dissemination is common, primary infection in adults involves lung tissue; Mtb interacts with the resident AMs (2, 10, 11). The Mtb has characteristic cell wall composition which hampers the success to treat TB with the actions of several antibiotics (12, 13). The mycobacterial cell wall architecture has been extensively studied since the 1960s (14, 15). The wall has a thick peptidoglycan-arabinogalactan complex, mycolic acid, capsular components including polysaccharides (e.g., α-glucan), lipids (e.g., PDIMs and PIMs), and proteins (e.g., GroEL2) (12, 16). Given the unusually lipid-rich cell wall, mycobacteria exhibit distinct staining characteristics and are known as acid-fast bacteria (17). These cell wall components of the outermost Mtb compartments interact with host cells to suppress or activate immune responses (16).
Stepwise Mtb adaptation and survival within host lungs, which mount major immune system-based attacks against infection, employ specific bacterial strategies using signal transduction systems, redox sensors, and secretion complexes (18, 19, 20). One such strategy, the two-component system (TCS), which is mostly exhibited by bacteria, is a phosphorelay signal transduction pathway that senses signals from histidine kinase and mounts an appropriate transcriptional response via a cognate response regulator (21, 22). Mtb harbors 12 pairs of TCSs that combat host immune system function after the establishment of an infection (22, 23). Each system produces proteins to construct a favorable granulomatous environment from the harsh conditions such as hypoxia, depleting energy sources, lowering the pH, and producing reactive oxygen species (ROS) or reactive nitrogen intermediates. Mtb thus persists for some time without growth and is not recognized by host immune sensors (8, 21, 24). For example, the DosS-DosR system, a well-known TCS of Mtb, plays a crucial role in Mtb survival under hypoxic conditions (25, 26). After O2 depletion, DosS senses the low O2 level and then activates DosR to upregulate the transcription of approximately 50 downstream genes, including hspX (Rv2031c), which encodes the chaperone alpha-crystallin and contributes to maintenance of the dormant state by stiffening its cell wall (26, 27). Recent studies have shown that the HspX protein may serve as a vaccine against TB (28, 29, 30).
Most recent attention has focused on drug-resistant TB, which imposes a major global burden through direct infection with drug-resistant Mtb or resistance acquired via Mtb genetic mutation triggered by adverse side-effects, or a failure to comply with the drug regimen during prolonged treatment (31, 32). In 2022, TB was recognized as the most common cause of airway drug-resistant epidemics in the past two centuries (1, 31, 33). Thus, there is an urgent need for the development of new and simple curative strategies, i.e., effective anti-mycobacterial drugs. Many reviews have highlighted the importance of new host-directed therapeutics for TB and the need to study the association between Mtb and the host immune system (7, 34, 35).
The host–Mtb interaction is characterized by granulomatous inflammation, which contains and localizes Mtb at the site of infection (36). A granuloma, first described by Rich in 1951, is the hallmark lesion of TB, walling off Mtb from the host environment (37, 38). A granulomatous lesion is an organized structure containing various immune cells and other types of cells, i.e., macrophages, lymphocytes, monocytes, neutrophils, and fibroblasts (39). Granuloma formation is initiated by the innate immune system, although CD4+ T lymphocytes play a major role in integration of the granulomatous structure (36, 39, 40). Although debate continues, the host granulomatous response to Mtb infection involves both protective and destructive granulomas (36). Protective granuloma formation is mediated by the cellular and humoral arms of the immune system to eradicate Mtb. Hypoxia within a granuloma stresses Mtb; cellular genes regulated by the hypoxia inducible factor 1 subunit alpha (HIF-1α) pathway are activated to clear intracellular Mtb, and HIF-1α serves as a marker of hypoxia within a granuloma. Previous studies reported correlations between TB progression and HIF-1α production in both TB patients and healthy controls (41). HIF-1α activates lactate dehydrogenase (LDH), which converts intracellular pyruvate from glycolysis into lactate (42). Earlier studies found increased LDH levels in the bronchoalveolar lavage fluid or sputum of patients with active TB (43, 44). Moreover, HIF-1α-depletion, which lacks HIF-1α-activated LDH-A, increased the level of intracellular pyruvate (compared to wild-type) in bone marrow-derived macrophages, thus facilitating Mtb growth using host-derived pyruvate as an energy source (45). However, if the host immune response is dysregulated, irreversible tissue damage can trigger necrosis, cavitation, and the spread of infected Mtb into lung parenchymal tissues (36, 46). Indeed, excessive pro-inflammatory responses may cause extensive tissue damage and the persistence of non-replicating bacilli. In this context, destruction of lung tissue extracellular matrix, principally by matrix metalloproteinases, triggers necrosis of TB lesions and unfavorable outcomes (46).
Innate immune responses
The front-line lung defenses include various cells in the immune system and barrier structures. Mtb primarily infects lung innate immune cells, exploiting them to create an environment that aids Mtb replication. Alveolar epithelial cells (AECs) are the first physical barrier that counters Mtb lung infection (47). Type 1 AECs are involved in gas exchange. Type 2 AECs exhibit immunomodulatory functions, secreting lactoferrin, defensins, cathelicidins, ROS, metalloproteinase, and surfactant proteins that enhance macrophagic phagocytosis and inhibit Mtb growth (48, 49, 50, 51). AECs recognize Mtb via bacterial pattern recognition receptors (PRRs) and then produce chemotactic cytokines, such as interferon (IFN)-, tumor necrosis factor (TNF)-α, and granulocyte–macrophage colony-stimulating factor (GM-CSF), which recruit immune cells. Mtb can invade AECs, creating a replication-conducive environment and thus crossing the mucosal barrier that initially inhibited dissemination.
Bacterial Toll-like receptor (TLR) signaling activates the nuclear factor kappa B (NF-κB) and MAPK pathways (50, 52). Mtb replication within AECs triggers cell necrosis; bacteria are released and infect surrounding cells (53). The endothelial cells (ECs) that line the vasculature facilitate immune cell transport during inflammation. ECs regulate metabolic homeostasis, vascular permeability, and hemodynamics (54). ECs recognize pathogens through PRRs on pathogen surfaces and then produce ROS, reactive nitrogen intermediates, and antimicrobial peptides (54). However, Mtb can enter ECs through Rab5 and Rab7, aiding bacterial dissemination through the bloodstream and lymphatic system (55, 56). The pathogen-associated molecular patterns of Mtb, such as phthiocerol dimycocerosates and mannosylated lipoarabinomannan (Man-LAM), promote recognition via EC mannose receptors and subsequent EC infection (57, 58). Vascular endothelial growth factor (VEGF), secreted by macrophages infected with Mtb expressing the RD1 virulence factor, promotes angiogenesis and facilitates bacterial spread (59). The serum VEGF levels of TB patients are elevated (60, 61, 62), and a VEGF-inhibitory therapeutic strategy has been used to normalize granuloma status and reduce bacterial dissemination (63, 64). Therefore, an understanding of the interaction between Mtb and the host is crucial to develop effective therapeutic strategies. This section focuses on the interactions between Mtb and innate immune cells, including macrophages, DCs, and neutrophils.
Macrophages
Macrophages play key roles in the immune response against Mtb via bacterial phagocytosis, initiation of inflammatory responses, and coordination with other immune cells. AMs and interstitial macrophages (IMs) play different roles during fetal lung development (65, 66). AMs maintain barrier integrity and regulate surfactant production, whereas IMs control the actions of T cells and DCs during infection (67). Mtb-infected macrophages undergo metabolic changes; AMs exhibit increased fatty acid oxidation, and IMs exhibit more glycolysis, affecting the ability of cells to control bacterial growth (68). Macrophages are classically activated by Th1 cytokines (M1 macrophages), which are pro-inflammatory and bactericidal; alternatively, they are activated by Th2 cytokines (M2 macrophages), which are anti-inflammatory and involved in tissue repair (69). Mtb effectors such as ESAT-6 drive M1-type polarization at an early stage but subsequently induce an M2-like anti-inflammatory phenotype in AMs to facilitate Mtb replication (70, 71). However, IMs exhibit a pro-inflammatory M1 phenotype, effectively restricting bacterial growth (71). Other Mtb proteins, such as RP105, Rv1987, PPE36, and EspC, influence macrophage activation and polarization via various intracellular pathways (72, 73, 74, 75).
Macrophages, major components of granulomas, play a critical role in Mtb death and contribute to immunopathology in a context-dependent manner. Granulomas contain Mtb infection but may also serve as niches within which bacteria can survive and replicate. Macrophages are involved in the initiation of granulomatous lesions and can develop into both epithelioid cells and multinucleated giant cells (76). Macrophages are recruited to sites of infection by the ESX-1/RD1 region of Mtb; they phagocytose the bacteria and then undergo apoptosis, which is associated with new macrophage recruitment and propagation of the infection (38). A computational model suggests that the macrophage polarization ratio predicts granuloma outcomes. M1 polarization early in infection and certain dynamics of the NF-κB response may provide a favorable outcome (77).
Macrophages produce a range of cytokines, including TNF-α, interleukin (IL)-1β, and IL-6, which recruit other immune cells and maintain the inflammatory environment required to control Mtb infection. Macrophages also interact with other immune cells to enhance antigen presentation and T cell activation, recruit neutrophils to sites of infection, and support B cell activation, thus bridging the gap between innate and adaptive immunity. An understanding of how macrophage polarization is regulated in infective microenvironments and the factors that control macrophage functions in granulomas is essential to determine the fate of granulomas and influence disease outcomes.
Macrophages employ several mechanisms to defend against Mtb, including autophagy, apoptosis, and pyroptosis (78). Autophagy aids elimination of intracellular pathogens, while apoptosis and pyroptosis are forms of cell death that also enhance Mtb removal by recruiting and activating new phagocytes (79, 80, 81). However, Mtb has evolved mechanisms that manipulate these processes to its own advantage; Mtb inhibits autophagy via upregulation of IL-10 and histone modification (82, 83). Mtb also employs various evasion strategies, including inhibition of phagosome maturation by the iron-sulfur cluster protein WhiB3 and regulation of non-coding RNA expression (78, 84). An understanding of these mechanisms will inform the development of new therapeutic strategies.
Neutrophils
Neutrophils, one of the most common immune cells in the airways of individuals with active TB, are also key players in the innate immune response against Mtb (85). Their primary function is phagocytosis; the cells engulf and attempt to kill Mtb using various agents such as ROS and antimicrobial peptides. Neutrophils secrete pro-inflammatory cytokines and chemokines, which recruit and activate other immune cells at a site of infection. Neutrophils can undergo apoptosis and be phagocytosed by macrophages, thus transferring their antimicrobial contents to the macrophages and aiding bacterial killing within the latter cells (86). Further, neutrophils interact with DCs to enhance antigen presentation and T cell activation, bridging innate and adaptive immunity. However, Mtb has evolved mechanisms that neutralize ROS, including the enhanced intracellular survival (eis) gene and a type I NADH dehydrogenase (NuoG) that inhibits ROS production and thus protects the bacteria (82, 87, 88). Additionally, Mtb induces neutrophil necrosis, such that Mtb survives and the infection spreads (89, 90). Mtb also resists neutrophil extracellular traps (NETs); these web-like structures are composed of DNA and antimicrobial proteins that immobilize and kill pathogens (91). Also, excessive neutrophil activity and NET production can trigger tissue damage and necrosis, impairing the ability to control Mtb infection (92, 93). Therefore, the development of therapies that target the evasive Mtb mechanisms might enhance the effectiveness of neutrophil-mediated Mtb killing.
DCs
DCs are essential antigen-presenting cells (APCs) that bridge the gap between the innate and adaptive immune systems. DCs process and present antigens to T cells via major histocompatibility complex (MHC) molecules, thus both initiating and modulating the adaptive immune response. DCs initially exist as immature cells below the lung epithelial layer, but when they encounter antigens they mature and migrate to lymph nodes to prime T cells. The Mtb NuoG protein delays DC migration and antigen uptake by inhibiting the apoptosis of infected neutrophils (87). Although DCs produce pro-inflammatory cytokines such as IL-12, which is essential for differentiation of T helper 1 (Th1) cells, the Man-LAM, ESX-1, and glycolipid Di-O-acyl trehalose of Mtb manipulate the protective function of DCs (5, 94, 95, 96). In addition, Mtb promotes Th17 cell expansion rather than expansion of IFN--producing CD4+ T cells (97). DCs also contribute to the formation and maintenance of the granulomas that isolate Mtb and suppress Mtb growth. However, granulomas also “hide” Mtb, protecting the bacteria from host aggression (98, 99). Therefore, further studies on DCs and their interactions with other immune cells during Mtb infection might provide valuable insights into the pathogenesis of TB and suggest potential therapeutic strategies.
Adaptive immune response
The adaptive immune response, particularly the roles played by T cells and B cells, is crucial to control Mtb infection and prevent TB progression. T cells directly kill infected cells and maintain granulomas; B cells produce antibodies and assist in T cell activation. Appropriate interplay between innate and adaptive immunity is vital to mount a comprehensive defense against Mtb. Upon exposure to Mtb, humans and mice require approximately 4–6 and 2–3 weeks, respectively, to develop antigen-specific T cell responses in lymph nodes. These delays highlight the critical role played by the innate immune response in the early stages of infection.
CD4+ T cells, also known as helper T cells, are essential for orchestration of the immune response against Mtb. These cells are activated by APCs that present Mtb antigens via MHC-II molecules. Once activated, CD4+ T cells produce cytokines, such as IFN- and TNF-α, which activate macrophages and enhance their bactericidal activities (100, 101). Cytokines are crucial for the formation and maintenance of granulomas, which contain the infection. Granulomas are composed of infected macrophages surrounded by T cells; granuloma integrity is vital to prevent Mtb spread (102, 103). CD8+ T cells, also known as cytotoxic T lymphocytes (CTLs), recognize and kill Mtb-infected cells after the T cells are activated by Mtb antigens presented via MHC-I molecules. CD8+ T cells release perforin and granzymes, which induce the apoptosis of infected cells and thereby limit the bacterial load. Like CD4+ T cells, CD8+ T cells contribute to granuloma formation and maintenance by secreting cytokines and exerting cytotoxic effects on infected macrophages (104). Both CD4+ and CD8+ T cells are involved in granuloma formation (105). CD8+ T cells from the blood of TB patients were more active than those from healthy controls (106). Regulatory T cells (Tregs) play a dual role in TB; these cells modulate the immune response to prevent excessive inflammation, which can lead to tissue damage. However, Mtb can cause Tregs to suppress the immune response (107, 108, 109). An understanding of the balance between effector T cell and Treg actions is crucial to develop therapies that enhance protective immunity without causing excessive tissue damage.
B cells produce antibodies that neutralize and thus aid the clearance of Mtb. Antibodies also opsonize bacteria, enhancing Mtb recognition and phagocytosis by macrophages. B cells contribute to long-term immunity and serve as APCs that present antigens to T cells. Recent studies have reported an Mtb-specific IgM reaction and Mtb-specific B cell responses in macaques and ex vivo human assays (110, 111). A better understanding of these mechanisms will further improve the vaccines and other therapeutic strategies that aim to control TB.
Autophagy restricts intracellular Mtb infection
In response to various stresses, autophagy plays a critical role in maintaining homeostasis. This lysosome-dependent catabolic process sequesters and degrades cytosolic macromolecular components (112). In early studies, macroautophagy (hereinafter autophagy) was viewed as a form of nonspecific degradation of damaged organelles and large macromolecules. Autophagy involves several consecutive steps, including phagophore formation, elongation into autophagosomes, and fusion thereof with lysosomes (113). The autophagy pathway (including macro- and micro-autophagy) is mediated by several autophagy-related genes (ATGs). Each ATG plays a critical role; sometimes, several ATGs combine to activate various autophagic processes (114). Of the various ATGs, the mammalian Atg8s play essential roles in both autophagy induction and lysosomal biogenesis by activating the EB transcription factor (115). Although many studies have explored the different functions of the ATGs that activate autophagy, more work is needed to fully understand the autophagy-dependent and -independent roles of various ATGs during infection.
In this section, we briefly discuss attempts to activate autophagy for elimination of intracellular Mtb, the use of xenophagy to control Mtb infection, and noncanonical autophagy during mycobacterial infection. Finally, we discuss several ways in which Mtb evades host autophagy, although we do not deal with all Mtb components that modulate host autophagy.
Activation of autophagy to control intracellular Mtb
Many exogenous stimuli, small molecules, and other agents have been reported to activate autophagy (Fig. 1), presumably nonselectively (116). An early study by Deretic’s group showed that IFN- promoted autophagic clearance of intracellular Mtb (117). Several PRRs, including TLR4 and TLR7, activate autophagy and thus promote the innate host defense (118, 119, 120, 121). In human monocytes/macrophages, vitamin D3 strongly induces autophagy and thus enhances intracellular Mtb control by LL-37 (an antimicrobial peptide) (122). A previous study found that FDA-approved drugs, such as carbamazepine and valproic acid, induced autophagic clearance of intracellular Mtb from primary human macrophages (123). Importantly, carbamazepine upregulated antimicrobial autophagic activities even against multidrug-resistant Mtb (123). Several natural products induced autophagy that suppressed intracellular Mtb survival in host cells. Natural marine clionamines enhanced autophagy and suppressed intracellular Mtb survival by targeting Pik1 (124). Many drugs and other agents improved antimicrobial activities by inducing autophagy (116), suggesting potential utility of autophagy-based adjunctive host-directed therapeutics for TB.
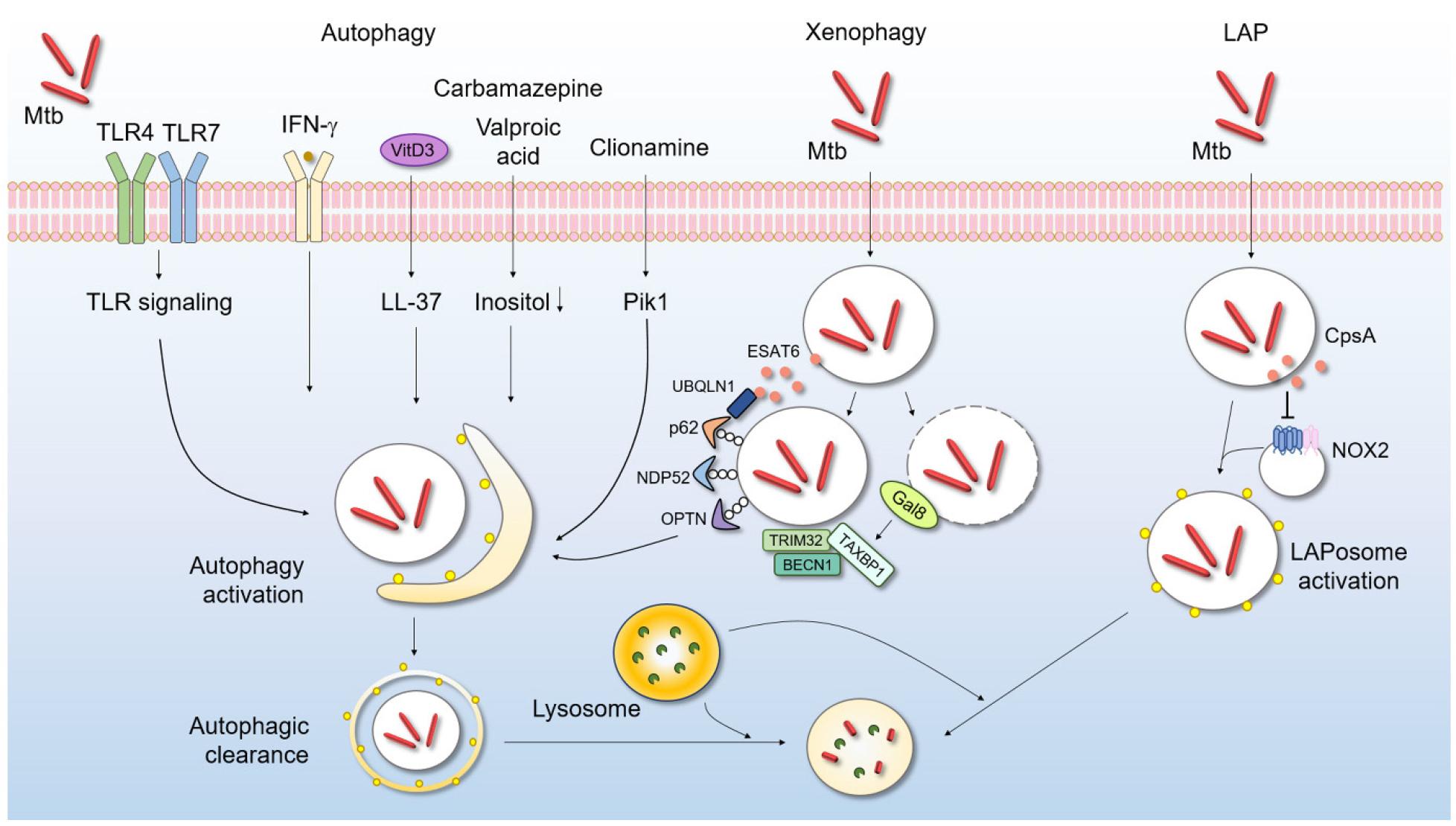
Fig. 1
Autophagy protects against Mtb infection. Autophagy is a key means by which a host eliminates intracellular bacteria, including Mtb. After Mtb phagocytosis occurs via various routes, autophagy is activated. Activated autophagosomes fuse with lysosomes to degrade and clear Mtb. Macroautophagy can be triggered by several extrinsic signals including PRR activation; IFN- generation; and treatment with vitamin D3, several drugs, or natural products. Xenophagy and LAP (distinct autophagic pathways) contribute to intracellular Mtb elimination. IFN, interferon; LAP, LC3-associated phagocytosis; LAPosome, LC3-associated phagosome; Mtb, Mycobacteriumtuberculosis; NOX2, NADPH oxygenase 2; ROS, reactive oxygen species; TLR, Toll-like receptor; VitD3, vitamin D3.
Targeting of xenophagy to combat Mtb infection
Recent studies have revealed selective forms of autophagy targeting different cargos and organelles; the forms include mitophagy, xenophagy, and reticulophagy (125). Xenophagy is a specialized form of autophagy that focuses on foreign organisms (126). Several autophagic receptors, including p62/SQSTM1, NDP52, and OPTN, are involved in target recognition, linkage to the autophagic machinery, and eradication of intracellular bacteria (127, 128, 129). In addition, galectin-8 senses phagosomal damage and then recruits TAX1BP1, a xenophagy receptor, to target Mtb and limit its intracellular replication (130).
Interestingly, the host protein ubiquilin-1 interacts with Mtb via ESAT-6/EsxA, thereby facilitating ubiquitin, p62, and microtubule-associated protein 1 light chain 3 (LC3) accumulation around Mtb and thus bacterial control (131). Additionally, the ubiquitin ligase TRIM32 enhances autophagic clearance of Mtb in macrophages by recruiting the autophagic proteins NDP52 and BECN1 (132). Chemical mimics of N-degron (Nt-Arg) activate xenophagy to enhance the host defense against multidrug-resistant Mtb and Salmonella both in vivo and in vitro(133). Although recent studies have highlighted the crucial role played by xenophagy activation in Mtb infection control, more work is required to determine whether these newly emerging drugs/agents could control drug-resistant TB.
LC3-associated phagocytosis during Mtb infection
As mentioned above, canonical autophagy is triggered by a variety of stress signals (such as nutrient deprivation) and is regulated by the induction of adenosine monophosphate (AMP)-activated kinase (AMPK) production (134, 135). LC3-associated phagocytosis (LAP) is a noncanonical autophagic pathway that differs in several ways from canonical autophagy. One unique characteristic of LAP is lipidation of LC3/Atg8 within single-membrane vesicles (136) via a process that requires RUBCN, a RUN-domain-containing protein that plays an inhibitory role during canonical autophagy (137). Another distinct feature of LAP is the production of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 (NOX2)-derived ROS during phagosome maturation (137). In addition, LAP operates independently of AMPK–mammalian target of rapamycin (mTOR) complex 1 (mTORC1) pathways (137).
Accumulating evidence suggests that, during various bacterial infections, LAP is activated upon recognition of PRRs and then contributes to the host defense against a variety of intracellular bacterial infections (135). In the Mtb context, CpsA, a LytR-CpsA-Psr domain-containing protein, aids Mtb evasion of LAP and thus reduces Mtb killing by NOX2 (138). However, our knowledge of noncanonical (compared to canonical) autophagy during Mtb infection is limited. As LAP is important in terms of immune regulation and dead cell removal in several pathogenic conditions (136), future studies are warranted to elucidate the Mtb LAP effectors and the molecular mechanisms by which LAP’s actions are orchestrated during a hyperinflammatory TB infection. Such efforts would aid the development of new therapeutic strategies that specifically modulate LAP during the different clinical stages of infection.
Mtb effectors of autophagic evasion
Mtb is a successful human pathogen that evades host autophagy/xenophagy using many different tactics (Table 1). For example, the eukaryotic-like serine/threonine protein kinase G (PknG; Rv0410c) of Mtb inhibits autophagosome maturation by interacting with RAB14, a small GTPase of the host, thereby promoting Mtb survival within host cells (139). Additionally, Mtb produces a terpenyl nucleoside, 1-tuberculosinyladenosine (1-TbAd; Rv3377c-Rv3378c), which blocks both autophagy and lysosomal maturation in association with lipid storage in foamy macrophages (140). The Mtb acyl carrier protein (Rv2244) suppresses the nuclear translocation of transcription factor of EB (TFEB) and phagolysosomal fusion within macrophages (141). The Mtb effector PPE51 (Rv3136) suppresses the TLR2 signaling response, particularly that of extracellular signal-regulated kinase 1/2 (ERK1/2), and inhibits autophagic activation (142). A previous study showed that PE_PGRS20 (Rv1068c) and PE_PGRS47 (Rv2741) blocked the initiation of canonical autophagy by interacting with the Ras-related protein Rab1A (143). Although we have not cited all research in this field, further studies are required to identify more Mtb effectors and the molecular mechanisms by which host canonical and noncanonical autophagy are finely regulated. These efforts will uncover the mechanisms by which Mtb survives in host cells during infection, and may suggest new therapeutic strategies.
Table 1.
Mtb effectors aiding autophagic evasion
| Effectors | Evasion strategy | Outcome | Ref. |
|---|---|---|---|
| PknG | Interaction with RAB14 | Inhibition of autophagosome maturation | (139) |
| 1-TbAd | Inhibition of autophagy and lysosomal maturation | Lipid storage in foamy macrophages | (140) |
| AcpM | Inhibition of TFEBa nuclear translocation | Suppression of phagolysosomal fusion | (141) |
| PPE51 | Inhibition of the TLRb signaling response | Inhibition of autophagic activation | (142) |
|
PE_PGRS20 PE_PGRS47 | Interaction with Rab1A | Blockage of canonical autophagy initiation | (143) |
Cytokine networks: Host protection, pathologies, and biomarkers
During Mtb infection, both macrophages and neutrophils, two sentinels of innate immunity, play critical roles in mounting a host defense via activation of cytokine and chemokine production and the oxidative burst (144). Although early activation of pro-inflammatory mediators is critical to combat intracellular Mtb in host cells, this also induces tissue damage (144). One review suggested that the key immune markers of different stages of TB infection included various cytokines, such as IL-1β, IL-6, IL-8, IL-10, and IL-12, as well as matrix metalloproteinases and inhibitors thereof (145). A recent study on the plasma biomarkers of TB patients revealed that distinct inflammatory cytokine profiles, including elevated BAFF, LIGHT, sTNF-R1 and sTNF-R2, and IP-10 levels, were associated with the clinical severity of TB (146). A systematic review found that the actions the cytokines IFN-, IL-2, and TNF-α had been most frequently investigated, although further well-designed studies are warranted (147). Therefore, many cytokines/chemokines may play critical functions in infective outcomes and might also serve as biomarkers of different infective TB stages, as well as the positive outcomes of adequate pharmacotherapy.
In this section, we discuss four major innate immune cytokines that have been widely studied in terms of their actions in human TB (Fig. 2).
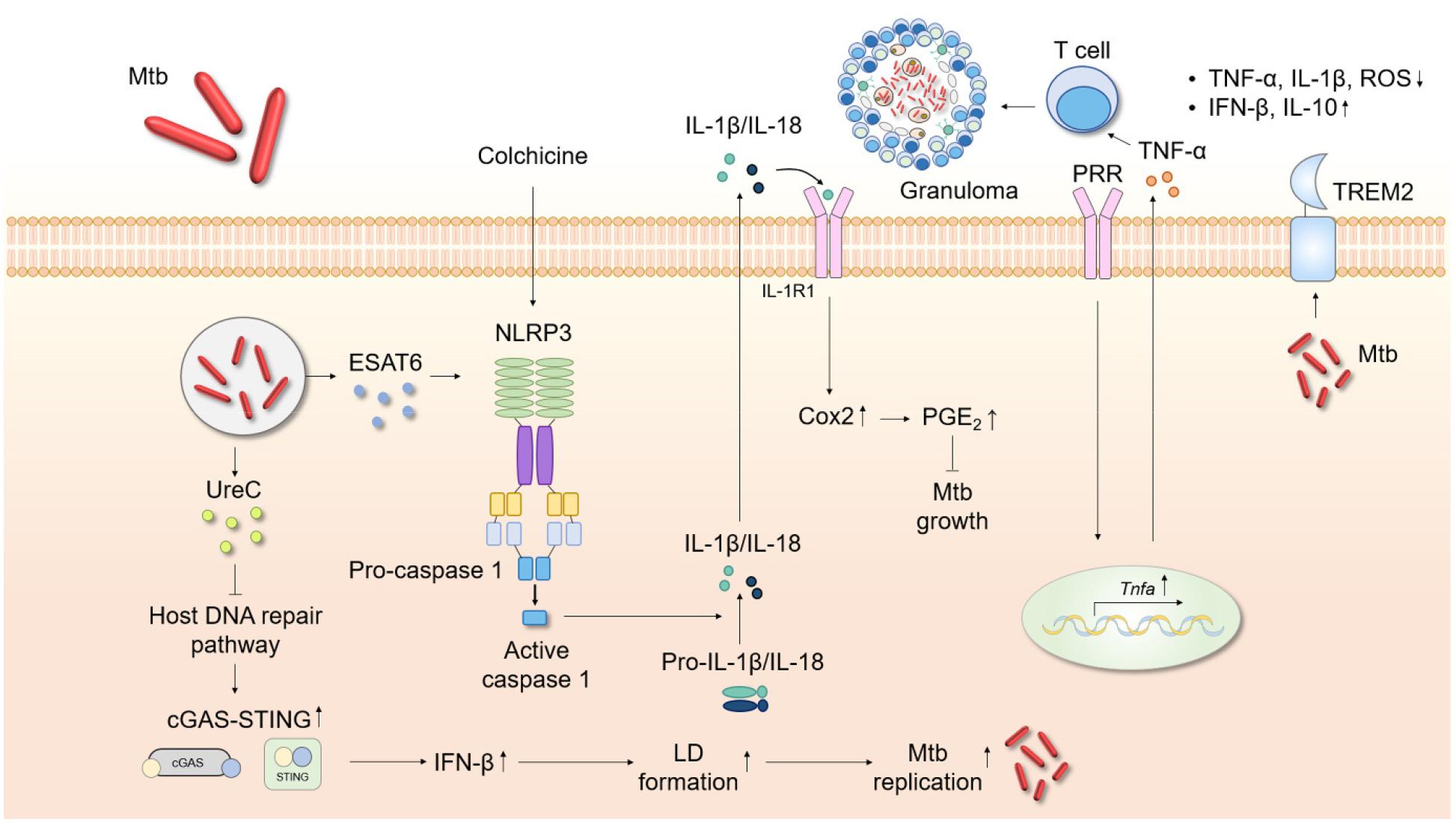
Fig. 2
Schematic showing the roles played by inflammatory cytokines during Mtb infection. Cytokines are crucial in host cell protection against Mtb infection. TNF-α upregulation recruits immune cells and stimulates granuloma formation. Mtb activates the NLRP3 inflammasome via ESAT6, followed by maturation of caspase-1, IL-1β, and IL-18. Colchicine-induced IL-1β production triggers Cox2/PGE2 signaling, suppressing Mtb growth. The Mtb protein UreC inhibits host DNA repair and thus activates cGAS-STING signaling. The level of IFN-β then increases, upregulating LD formation and facilitating Mtb replication. TREM2 blocks TNF-α, IL-1β, and ROS generation and promotes IFN-β and IL-10 synthesis. cGAS-STING, cyclic GMP-AMP synthase/stimulator of interferon genes; Cox2, cyclooxygenase 2; IFN, interferon; LD, lipid droplet; Mtb, Mycobacterium tuberculosis; PGE2, prostaglandin 2; PRR, pattern recognition receptor; TREM2, triggering receptor expressed on myeloid cells 2.
TNF-α
TNF-α is one of the most important cytokines in terms of TB progression. TNF-α is involved in M1 macrophage activation, apoptosis induction and regulation, and immune cell recruitment (148, 149). Also, TNF-α plays a critical role in host protection via granuloma formation and immune cell recruitment (148, 150). Anti-TNF therapy for patients with autoimmune diseases often predisposes them to an increased risk of TB re-activation (151). Such findings strongly suggest that TNF-α aids the host defense against TB. However, excessive TNF-α production leads to pathological responses, including cachexia, which is a major adverse event in humans with TB (148). Therefore, appropriate TNF-α production is required to maintain the host immune defense, without any toxic effects, during Mtb infection.
In patients with both pulmonary and intracerebral Mtb infections, TNF-α is critical in terms of resistance (150, 152). Infants are more susceptible to TB infection than older cases because several important cytokines, including TNF-α and IL-1, are produced only at low levels, and precursor cell differentiation into IFN--producing T cells (153) is thus minimal. More studies are needed to understand the multifaceted roles played by TNF-α at different stages of various mycobacterial infections.
IL-1
Secretion of IL-1 (including IL-1β and IL-18) is modulated by inflammasome activation of innate immune cells. The inflammasome complex is a protein assembly with a sensor, an adaptor, and a caspase that is triggered by inflammatory signals. This multimeric protein platform activating caspase-1 is also responsible for the maturation of IL-1β and IL-18 (154). Of the many inflammasome complexes, the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome has been the most widely studied.
Previous studies examined Mtb-manipulated, IL-1-derived pathogenic responses. The Mtb protein ESAT-6 triggered NLRP3 inflammasome activation in different cell types, leading to caspase-1 maturation and IL-1β release (155, 156). An Mtb ESAT-6 mutation was associated with no increase in the IL-1β or IL-18 level (157). Prolonged elevation of IL-1 levels during Mtb infection can damage tissue (158, 159). By modulating the IL-1β concentration, Mtb ensures its own survival and increases disease development associated with necrotic cell death (160, 161). The vaccine strain M. bovis Bacillus Calmette–Guérin (BCG) does not activate NLRP3 inflammasome assembly. Gain-of-function genetic changes in the NLRP3 gene of human macrophages increase Mtb growth restriction in such macrophages (162). Based on the pathogenic role played by IL-1 in TB, novel preventative strategies have been developed. TB anti-IL-1 therapy (IL-1 neutralizing antibodies) controlled inflammation by mitigating IL-1 signaling (158, 163, 164). However, such immunosuppressive treatment enhances the susceptibility of TB patients; a therapy that promotes IL-1 pathway activity without inducing a cytokine storm may be needed to avoid TB progression (165, 166). A recent study revealed that IL-1 blockade alleviated side effects associated with anti-TB linezolid treatment (159).
An early study showed that IL-1β production was required to protect against Mtb infection, regardless of the caspase-1 level (167). Mtb infection of mice lacking the Il1a, Il1b, or even genes of IL-1 receptor type 1 (IL-1R1) was associated with higher Mtb loads and severe lung pathology (168, 169, 170). Blockade of the adaptor myeloid differentiation factor 88 was also associated with high mortality after Mtb infection, reflecting dysfunctional IL-1R signaling (171). Furthermore, IL-1 plays a critical role in host resistance against Mtb infection by inducing eicosanoids via suppression of type I IFN synthesis (172). Another recent study showed that colchicine, a widely used inhibitor of NLRP3 inflammasome activation, rather induced Nlrp3 and Il1b gene expression. The secreted IL-1β increased IL-1R1 expression, and that of cyclooxygenase-2 (Cox2) and the lipid immune mediator prostaglandin E, effects associated with anti-Mtb actions (173). Additionally, Mtb promoted NLRP3-independent IL-1β release in lung tissues (174). Together, these data suggest that fine control of IL-1 signaling and inflammasome activation may aid the development of host-specific therapeutics.
IL-6
IL-6 is a pleiotropic cytokine that plays diverse roles in T and B cell regulation, and pro- and anti-inflammatory responses, in a context-dependent manner (175, 176). Thus, IL-6 is critical for the induction of chronic inflammation and the host defense against a variety of infections (176, 177). Recent studies have suggested that IL-6 may serve as a biomarker of the various infectious stages of human TB. Importantly, patient studies indicated that the pre-treatment IL-6 level may be a biomarker of unfavorable outcomes and disease progression during TB treatment (178, 179). An increased plasma IL-6 level was associated with a higher risk of lung cavitation in patients with drug-resistant TB (180).
Moreover, IL-6 production caused by the Mtb protein Rv0183 (a lysophospholipase) was higher in the whole blood of patients with active pulmonary TB than controls, suggesting that the protein might serve as an immunodiagnostic biomarker (181). A recent study showed that both IL-6 and IL-27 were potentially useful biomarkers of human pleural TB infections (182). In patients with tuberculous meningitis, the IL-6 level appears to be upregulated, and it may thus be a valuable biomarker (183).
However, IL-6 is a double-edged sword. Mtb inhibits host regulation of IL-6 production to suppress the activation of type I IFN pathways, thus exacerbating the disease (184). IL-6 suppressed the autophagic pathway activity promoted by IFN- by counteracting the protective effect of IFN- in Mtb-infected macrophages, thus increasing Mtb survival (185). Moreover, Mtb uses the ESAT-6 protein to upregulate IL-6 production, in turn activating the signal transducere and activator of transcription 3 (STAT3) protein; the action of this transcription factor is enhanced by multiple cytokines, including IL-6, in both mouse and human macrophage Mtb infections (186, 187).
IL-6 plays important protective roles against Mtb infection. Early studies showed that Mtb infection of mice with a genetic IL-6 deficiency increased the bacterial loads (compared to those of wild-type mice) by changing the responses of type I T helper cells (188, 189, 190). Additionally, IL-6 induced and maintained a well-controlled Th17 immune response, which has been suggested to be crucial to protect against TB (191). Moreover, IL-6 was required to prime IFN--secreting T cells during TB vaccination of Il6-knockout mice or mice in which IL-6 was specifically neutralized; such mice were otherwise not well-protected by vaccination (192). IL-6 is also associated with differentiation of CXCR5+ T follicular helper cells; IL-6 mediates appropriate polarization of T follicular helper cells to macrophages containing Mtb, which optimizes the anti-mycobacterial response (193). Future studies should determine whether IL-6 bioactivity might serve as a clinically predictive biomarker in various TB infection settings.
Type I IFNs: IFN-α and IFN-β
Excessive production of type I IFN during TB infection potentiates disease pathogenesis and exacerbation (172). An important question concerns how type I IFN is produced during infection. A recent study showed that Mtb urease C (UreC, Rv1850) suppressed the host DNA repair pathway and thus activated the cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS)/stimulator of IFN genes (STING) pathway. The resultant activation of IFN-β production facilitated lipid droplet (LD) formation, thus contributing to Mtb replication (194). Additionally, transcriptome analysis of M. marinum-infected animal models showed that type I IFN-associated pathways were enriched in an inoculum-dose-dependent manner and became activated at high multiplicities of infection (195). Mtb carrying a rifampicin drug-resistance-conferring single-nucleotide polymorphism (SNP) in the RNA polymerase gene H445Y SNP inhibited lung myeloid and lymphoid immune responses via type I IFN-dependent signaling pathways to a greater extent than did wild-type Mtb (196). These data suggest that the bacterium itself, and its heterogeneity, are strongly associated with type I IFN production.
Several host factors activate type I IFN production during Mtb infection. Triggering receptor expressed on myeloid cells 2 (TREM2), a transmembrane surface receptor, suppressed TNF-α, IL-1β, and ROS production but increased the production of IFN-β and IL-10. Mtb binds to TREM2, rendering the intracellular conditions more favorable for bacterial survival via continuation of type I IFN signaling, thereby suppressing macrophage antimicrobial responses (197). Another recent study showed that IRGM1 deficiency increased IRGM3-dependent type I IFN signaling, suppressing Mtb-specific T cell expansion. Irgm1-deficient mice cannot control Mtb infections of neutrophils and AMs (198). Another recent study revealed that type I IFN, produced by plasmacytoid DCs derived via the formation of NETs, is released when control of Mtb replication fails and thus contributes to TB pathogenesis (199). These studies strongly suggest that type I IFNs aid bacterial survival and replication in lung tissues by inhibiting appropriate (protective) T cell helper 1 responses during infection.
Certain protective roles played by type I IFN during Mtb infection were apparent under specific conditions (200). Case reports showed that administration of both IFN-α and an antimycobacterial drug combination exhibited protective effects in patients with mycobacterial infections who responded poorly to the conventional drug regimen or suffered disease recurrence (201). Type I IFN exerted beneficial effects in the absence of IFN- signaling both in vivo and in vitro. Young patients struggling to control mycobacterial infections because of a lack of IFN- pathway activity improved after combined administration of IFN-α and antimycobacterial chemotherapy (202). In Mtb-infected mouse models, type I IFN is required for blockade of the early pulmonary pathology and mortality in the absence of IFN- receptors. Moreover, type I IFN enhanced BCG vaccine efficacy against Mtb infection by increasing the production of protective cytokines including TNF-α, IL-12, and IFN-(203, 204). Another study found that a basal level of type I IFN signaling was required for optimal generation of TNF-α and IL-12 by macrophages and DCs that counter Mtb infection (205, 206).
Further studies are required to identify the key cytokines during TB infection and where they function within the innate immune regulatory network. Do they protect the host, and could they serve as diagnostic and/or predictive markers?
CONCLUSION
Balanced regulation of the host innate immune and inflammatory responses is critical to protect the host against TB. A granuloma is the hallmark of TB. Protective granuloma formation is critical to ensure immunometabolic limitation of bacterial proliferation at infection sites. Dysregulation of granuloma formation leads to Mtb spread. Future studies should explore how protective granulomas are maintained and the factors involved in TB persistence and progression.
It is clear that the proinflammatory cytokine TNF-α plays critical roles in granuloma formation and the development of protective immunity against TB infection. However, excessive TNF-α production triggers pathological inflammation and cell death. IL-1 is another protective cytokine produced via inflammasome activation. IL-6 is a pleiotropic cytokine that has recently served as a diagnostic and predictive marker in various stages of TB infection. Finally, the actions of pathological type I IFNs during TB infections have been widely studied. It may be that no single cytokine or chemokine can serve as a biomarker of disease progression or protection. Future studies should explore the cytokine networks that finely tune the protective and pathological immune responses.
Both canonical and noncanonical autophagy are now being recognized as crucial immune system contributors to the progression/control of mycobacterial infections. However, further preclinical and clinical studies are required to develop different autophagy-based adjunctive therapeutics for patients with drug-resistant infections and those in different stages of TB infection, and more mechanistic studies are needed to elucidate the Mtb effector–host interactions that regulate autophagy and other defensive pathways. More extensive investigations of host–pathogen interactions are required to develop more efficacious adjunctive treatment modalities. We must move to a host-directed therapeutic approach.
ABBREVIATIONS
AM, alveolar macrophage; AEC, alveolar epithelial cell; AMP, adenosine monophosphate; AMPK, adenosine monophosphate kinase; APC, antigen presenting cell; ATG, Autophagy-related gene; BCG, bacillus Calmette- Guérin; cGAS/STING, cyclic GMP-AMP synthase/stimulator of interferon genes; Cox2, cyclooxygenase-2; CTL, cytotoxic T lymphocyte; DC, dendritic cell; EC, epithelial cell; ERK1/2, extracellular signal-regulated kinase 1/2; ESX-1, ESAT-6 secretion system; GM-CSF, granulocyte-marophage colony stimulating factor; GMP, guanosine monophosphate; HIF, hypoxia-inducible factor; IFN, interferon; IL, interleukin; IL-1R1, interleukin 1 receptor, type I; IM, interstitial macrophage; LAP, LC3-associated phagocytosis; LC3, microtubule-associated protein 1 light chain 3; LD, lipid droplet; LDH, lactate dehydrogenase; Man-LAM, mannosylated lipoarabinomannan; MHC, major histocompatibility complex; Mtb, Mycobacteriumtuberculosis; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; NADPH, nicotinamide adenine dinucleotide phosphate; NET, neutrophil extracellular trap; NF-κB, nuclear factor kappa B; NLRP3, NOD-, LRR-, and pyrin domain-containing protein 3; NOX2, NADPH oxidase-2; PRR, pattern recognition receptor; RD, region of difference; ROS, reactive oxygen species; SNP, small nucleotide polymorphism; STAT3, signal transducer and activator of transcription 3; TB, tuberculosis; 1-TbAd. 1-tuberculosinyladonosine; TCS, two-component system; TFEB, transcription factor EB; Th1, T helper 11; TLR, Toll-like receptor; TNF, tumor necrosis factor; Treg, regulatory T cell; TREM2, triggering receptor expressed on myeloid cells 2; VEGF, vascular endothelial growing factor.
