

OVERVIEW OF MYCOBACTERIAL PATHOGENESIS
Mycobacterial spp., numbering over 190 within the Actinomycetota phylum (1), are obligate aerobes with GC-rich genomes that typically lack classical mobile plasmids and virulence factors (2, 3, 4). They are widespread in soil and water and exhibit physiological diversity: generally straight or curved rod-shaped, measuring 0.2 - 0.6 µm in width and 1.0 - 10 µm in length, and typically non-motile and non- spore-forming (5, 6). A hallmark of mycobacteria is their waxy, lipid-rich cell wall (Fig. 1), which yields an acid-fast characteristic and places them outside the classic Gram-positive/Gram-negative dichotomy; this property provides a useful diagnostic feature (7, 8, 9). The cell wall is thick and complex, consisting of a high concentration of mycolic acids - long-chain fatty acids (FAs) integral to the envelope and peptidoglycan (Fig. 1) (10, 11). This lipid-rich layer confers impermeability and intrinsic resistance to many antibiotics and environmental stresses (12, 13). High hydrophobicity further promotes growth in aggregates and branching forms (14, 15), facilitating surface adherence and biofilm formation, which contribute to intrinsic drug resistance (16).
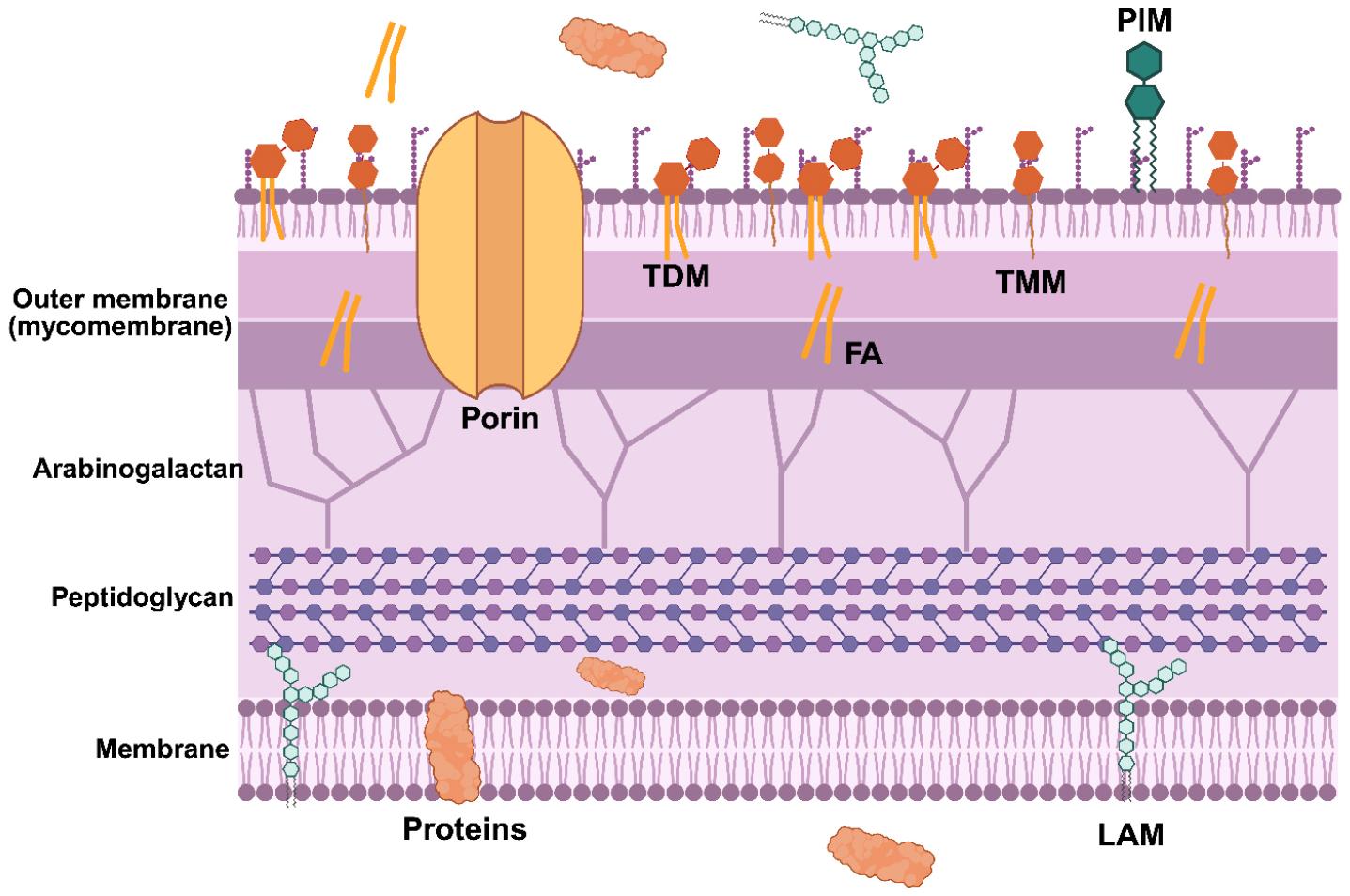
Fig. 1
Schematic representation of the Mtb cell envelope. The Mtb cell envelope is a multilayered architecture. The outermost mycomembrane consists of phospholipids and abundant trehalose-containing glycolipids - trehalose dimycolate (TDM) and trehalose monomycolate (TMM) - as well as other long-chain fatty acids, that form a highly hydrophobic barrier. Phosphatidyl-myo-inositol mannoside (PIM) and porin proteins embedded in the mycomembrane regulate permeability. Beneath the mycomembrane, the arabinogalactan–peptidoglycan complex forms a rigid cell wall core that is covalently linked to the mycolic acids. The innermost plasma membrane is composed of phospholipids, integral membrane proteins, and lipoarabinomannan (LAM). Created with BioRender.com.
Mycobacteria are generally categorized into three groups: the Mycobacterium tuberculosis complex, Mycobacterium leprae (the leprosy pathogen), and nontuberculous mycobacteria (NTM) (17, 18). A subset has evolved as opportunistic intracellular human pathogens, creating clinical relevance in both chronic pulmonary and non-pulmonary diseases (19, 20). Mycobacterium tuberculosis (Mtb),a representative member of the Mycobacterium tuberculosis complex, is a major human pathogen responsible for tuberculosis (TB) worldwide and the leading single-bacterial-agent cause of death (19, 20, 21). In 2024, an estimated 10.7 million TB cases and 1.3 million TB-related deaths were reported, with approximately 150,000 individuals coinfected with human immunodeficiency virus (HIV) (22).
The COVID-19 pandemic redirected funding and treatment toward COVID-19, creating gaps in TB diagnostics and care. Service limitations during this period were associated with fluctuations in TB incidence and mortality from 2020 to 2022 compared with preceding years; TB incidence has begun rising again, jeopardizing the WHO goal of an 80% reduction in new cases by 2030 (22). Although only about 5 - 10% of individuals infected with Mtb progress to symptomatic TB disease, including active and latent TB (Global Tuberculosis Report, WHO, 2025), the gradual increase in TB cases and deaths underscores the substantial clinical burden of these infections. Mtbpredominantly presents as pulmonary TB, given that transmission occurs mainly through airborne droplets from individuals with active disease (23, 24).
The virulence strategies by which Mtb evades host immunity remain incompletely understood. Mtbinvades via the airway, and alveolar macrophages are the first immune cells to engulf the bacteria and attempt degradation within the phagolysosome (25, 26). However, Mtb can counteract this by suppressing phagosome-lysosome fusion, thereby creating a favorable intracellular niche (27, 28). Well-known virulence factors unique to Mtb infection include early secreted antigenic target 6 kDa (ESAT-6), secreted through the ESX/type VII secretion system, which ruptures the phagosomal membrane prior to fusion with lysosomes and enables cell-to-cell spread (29, 30, 31).
The current first-line regimen for drug-susceptible TB comprises isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (ETH), administered for the first two months, followed by INH and RIF for an additional four months (32, 33). Despite decades of effort to eradicate TB, several obstacles impede successful treatment: i) Mtb can reside within host macrophages in latent states (e.g., dormant and persistent) after initial infection, complicating early diagnosis (34, 35); ii) the emergence of drug-resistant TB from poor adherence to prolonged regimen and the use of multiple antibiotics (36, 37); and iii) although the bacillus can resist the host immune response, few toxins or virulence factors have been identified, complicating vaccine, antibiotics, and diagnostics development (38, 39, 40). Consequently, a deeper investigation of Mtbadaptive strategies and metabolism required for persisting against host defenses and antibiotic effects is needed to identify new treatment strategies, including antibiotics and diagnostic candidates.
LATENT TB INFECTION
Persistent TB is a stable yet reversible physiological state in which Mtb markedly alters metabolic activity and networks while remaining viable, either non-replicating or replicating very slowly (41, 42, 43, 44, 45). This state features transient metabolic remodeling that allows Mtb to survive under adverse conditions and evade immune surveillance (46, 47). Latent TB infection is evolutionarily conserved and crucial for Mtb’s long-term survival within hosts and its pathogenicity, enabling persistence in susceptible individuals and evasion of clearance by the host immune system (48). Biologically, latent TB infection contributes to immune evasion, enduring persistence, antibiotic tolerance, and the formation of persister reservoirs (49). The large population of latently infected individuals underpins TB status as a global health burden (50). Latent bacteria can reactivate when host immunity or antibiotic pressure wanes, leading to active disease and complicating treatment with potential multidrug resistance and prolonged therapy (51, 52).
The hostile immunological microenvironment encountered by Mtb comprises diverse stressors regulated by both innate and adaptive immune components. This milieu includes coordinated release of immunoregulatory mediators - cytokines, chemokines, and interleukins - along with enhanced phagocytic activity and accelerated antigen presentation (53, 54, 55, 56). These immune responses create conditions that challenge bacterial survival and promote dormancy. Through dynamic interplay between host defenses and bacterial countermeasures, the tissue microenvironment within Mtb infection becomes increasingly harsh, ultimately promoting latent TB infection. Consequently, the host immune system’s efforts to eliminate bacilli actively contribute to Mtb latency. Phagocytosis by macrophages, followed by acidic, oxidative, and nitrosative stresses, as well as cytokine production, further constrains the intracellular environment (53, 57, 58, 59). Granuloma formation also occurs, establishing localized hypoxic and nutrient-poor microenvironments (60).
During infection, Mtb is exposed to multiple environmental and host-derived stressors that induce latency. Among these, hypoxia - defined as insufficient oxygen supply - serves as a primary signal for persistence in latent TB infection, reflecting conditions in the cores of lung granulomas over extended periods (51, 61, 62). To sense and adapt to low-oxygen environments, Mtb utilizes the heme-containing sensor kinases DosS and DosT in conjunction with the DosR regulon, forming a two-component signaling system (51, 63). Upon activation, the DosR regulon drives altered gene expression, initiating a dramatic metabolic remodeling from aerobic respiration to anaerobic or microaerophilic states (61).
Within the host, particularly in macrophages and granulomas, Mtb often encounters limited access to essential carbon and nitrogen sources. This nutrient limitation acts as another cue promoting metabolic adaptation for survival (64, 65). Consequently, Mtb shifts its central carbon and nitrogen metabolism, enabling utilization of alternative carbon sources such as fatty acids via the glyoxylate shunt and, when appropriate, the methyl citrate cycle (MCC) pathway (66). This metabolic flexibility supports energy production for long-term survival, with only modest reductions in viability during extended nutrient starvation (51, 67, 68).
To bolster antimicrobial defenses, host immune cells generate reactive oxygen species (ROS) and reactive nitrogen intermediates as a primary attack (69, 70, 71). In countermeasure, Mtb deploys adaptive strategies that include antioxidant enzymes (e.g., KatG, and superoxide dismutases) and cell-wall components (phthiocerol dimycocerosate, mycolic acids, and lipoarabinomannan) to counteract host pressures (51, 72, 73, 74). In this context, WhiB3 helps regulate cytoplasmic redox potential, protecting against molecular damage and supporting latency physiology (75, 76, 77).
As Mtb transitions into latency, its metabolic rate slows, cell division diminishes or ceases, and the bacterium enters a non-replicating state (78). This shift is accompanied by increased expression of genes associated with non-replicating persistence and decreased expression of metabolic genes (68). Upregulation of hypoxia-responsive genes and transcriptional adaptations to limited oxygen and nutrients further reinforce the latent state (79). Additionally, Mtb undergoes cell-wall remodeling and alterations in central carbon and nitrogen metabolism contributing to phenotypic antibiotic tolerance and improved survival under hostile conditions (64, 80).
CENTRAL CARBON METABOLISM ALTERED IN MTB PERSISTERS
Metabolomics is the holistic study of metabolic networks within an organism, including Mtb (81, 82). Understanding the metabolic adaptations that underlie Mtb’s transition to latent TB infection is crucial. The following subsections delineate four core metabolic strategies employed by Mtb persisters during latent TB infection to survive hostile host environments. These strategies also hold promise for developing metabolic diagnostic biomarkers to distinguish latent TB infection from active TB.
A. Cell-wall trehalose mycolates lysis: Trehalose-catalytic shift (Fig. 2)
The cell envelope of Mtb is among the most complex lipid-rich structures in nature (Fig. 1), contributing to its divergence from classical Gram-positive and Gram-negative bacterial boundaries. Understanding the structure and function of the Mtb cell wall is essential for elucidating its role in pathogenesis, including drug resistance, protection against host-derived stresses, and survival within granulomas. Mycolic acids – long-chain fatty acids – comprise a substantial fraction of the mycobacterial biomass, and trehalose-based glycolipids define key features of the mycomembrane (83, 84, 85, 86, 87).
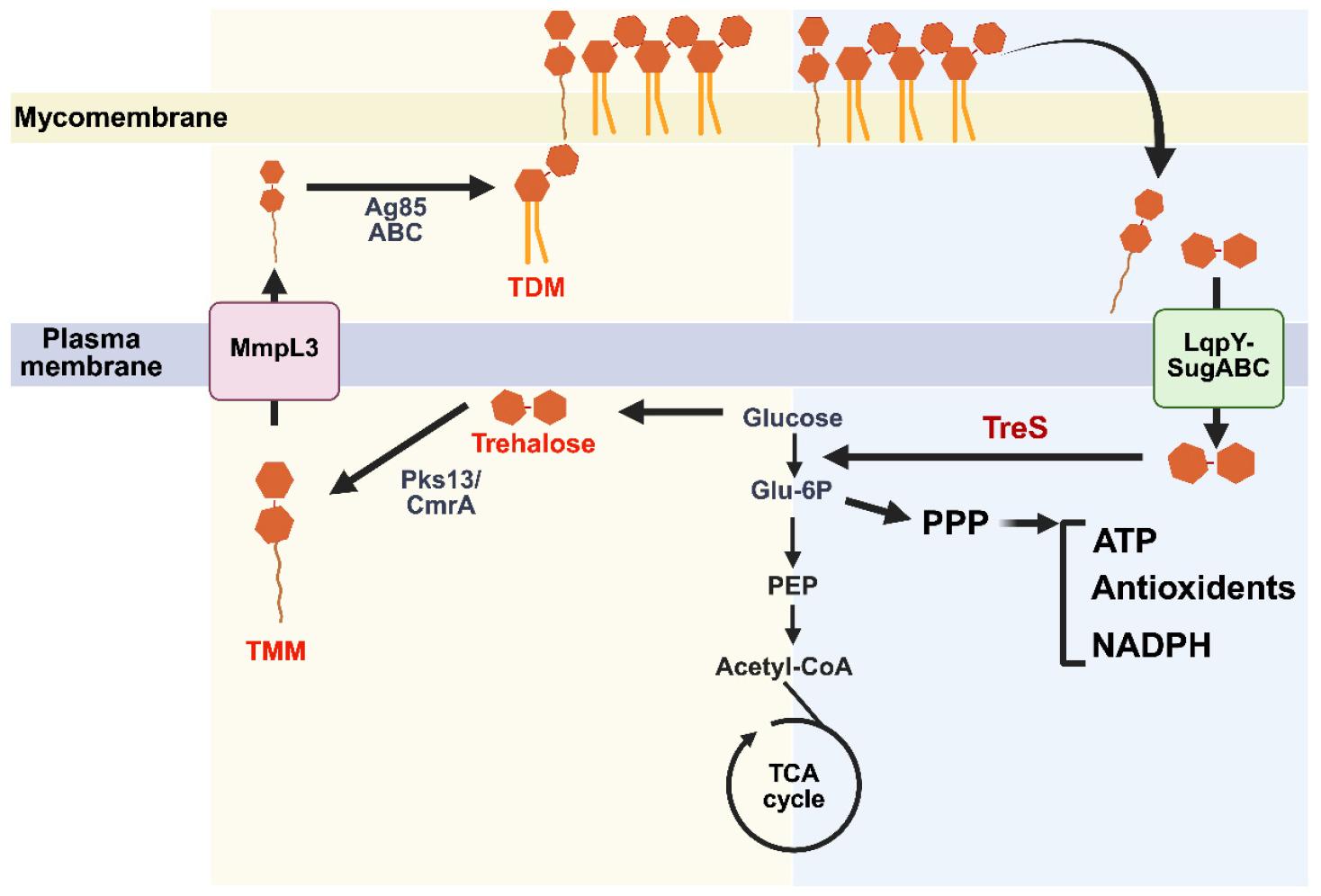
Fig. 2
Remodeling of cell wall trehalose mycolates linked to trehalose catalytic shift in Mtb. The schematic in the left yellow panel shows how trehalose-based glycolipids are synthesized, transported, and recycled in Mtb envelope. Trehalose monomycolate (TMM) is exported across the plasma membrane by the transporter MmpL3, where it serves as a precursor for trehalose dimycolate (TDM) formation in the mycomembrane by Ag85 (ABC) mycolyltransferases and for other trehalose-containing lipids catalyzed by Pks13/CmrA. Right blue panel, surface-exposed TMM/TDM can be hydrolyzed, and the released trehalose is re-imported into the cytoplasm through the LpqY–SugABC transporter complex, replenishing intracellular trehalose. Cytosolic trehalose is metabolized by trehalose synthase (TreS), feeding upper glycolysis and the pentose phosphate pathway (PPP) to sustain levels of ATP, NADPH, and antioxidants for Mtb persister physiology. Glu-6P: Glucose 6 phosphate; PEP: phosphoenolpyruvate. Created with BioRender.com.
Trehalose, a non-reducing disaccharide of two glucose units, plays multiple structural and metabolic roles in Mtb. Its de novo biosynthesis proceeds through three major pathways: OtsA/OtsB2, TreY/TreZ, and TreS (88, 89, 90). Within the cell envelope, trehalose is conjugated to long-chain mycolic acids to form trehalose monomycolate (TMM) and trehalose dimycolate (TDM), two signature glycolipids essential for cell-wall integrity, interaction with host immunity, and virulence (Fig. 1) (91, 92). TDM, also known as cord factor, engages the host monocyte-inducible C-type lectin receptor (Mincle), triggering pro-inflammatory cytokine responses and granuloma formation (93, 94, 95). Beyond immunomodulation, TDM inhibits phagosome–lysosome fusion, enabling intracellular survival (96, 97).
Trehalose-derived glycolipids underscore trehalose’s central role in Mtb physiology (98). Recent metabolomics and biochemical studies have redefined trehalose not only as a structural building block but also as a metabolic switch that links cell-wall degradation pathways to central carbon metabolism (CCM) (99, 100, 101). Under nutrient starvation, antibiotic exposure, or hypoxic stress, Mtbreroutes trehalose catabolism from glycolipid biosynthesis toward energy production and redox-balancing pathways, a process termed the trehalose-catalytic shift (Fig. 2) (100, 102, 103). This shift is regulated by trehalose synthase (TreS), which converts trehalose to maltose, which is then converted to glucose-6-phosphate (G6P). Redirected G6P fuels glycolysis and the pentose phosphate pathway (PPP), supplying substrates for energy production and redox balance to sustain survival under hostile conditions. Metabolomic profiling of Mtb from in vitro biofilms indicates that TreS activity drives metabolic reprogramming associated with persister formation. TreS-driven Mtb subpopulations exhibit enhanced tolerance to front-line TB antibiotics, including INH and RIF, as well as bedaquiline (BDQ); TreS-deficient strains show diminished persister formation and increased antibiotic susceptibility. Thus, TreS acts as a pivotal mediator of phenotypic heterogeneity, enabling subsets of Mtb to withstand otherwise lethal stresses (100, 103).
Intriguingly, this trehalose-catalytic shift appears more pronounced in drug-resistant (DR) TB clinical isolates than in drug-sensitive (DS) strains. Comparative metabolomics reveal elevated trehalose and catabolic intermediates, such as G6P, pentose-5-phosphate, and sedoheptulose-7-phosphate in DR-TB isolates relative to DS-TB isolates, indicating greater trehalose-to-CCM flux in DR-TB (103). This rerouting provides both bioenergetic resilience and redox buffering - traits that may facilitate emergence and maintenance of drug resistance. The correlation between TreS activity and drug-resistance phenotypes suggests that the trehalose-catalytic shift supports survival under antibiotic pressure and may foster mutagenic conditions conducive to acquiring genetic resistance (103).
To interrogate metabolic heterogeneity at the single-cell level, Swarts and colleagues developed a fluorogenic trehalose analogue, Red Molecular Rotor-trehalose (RMR-tre), which serves as a substrate for the Ag85 complex responsible for TDM biosynthesis (104). When added to Mtb cultures, RMR-tre is incorporated into the cell wall, yielding fluorescent signal proportional to TreS-associated trehalose-catalytic shift activity. Cells with high TreS activity divert endogenous trehalose toward CCM, resulting in reduced intracellular trehalose for TDM synthesis and greater incorporation of exogenous RMR-tre into TDM. TreS-deficient or low-activity cells rely predominantly on endogenous trehalose and display lower fluorescence (103).
Flow cytometric analysis of RMR-tre - stained populations reveals striking heterogeneity, even among genetically identical cells. Antibiotic-exposed cultures show a higher proportion of highly labeled subpopulations, corresponding to cells with elevated TreS activity and metabolic remodeling consistent with persisters. Drug-resistant Mtb exhibits a greater baseline fraction of high-labeled cells than drug-sensitive strains, suggesting that metabolic heterogeneity is a pre-existing trait within resistant populations. After drug exposure, the low-labeled fraction is selectively depleted, enriching the high-labeled survivors that may seed future resistant clones (103).
The RMR-tre labeling assay provides a powerful, non-genetic tool to visualize and quantify metabolic heterogeneity linked to persistence and resistance in Mtb (103, 105). Coupled with flow cytometry or high-content microscopy, this probe enables dynamic tracking of metabolic states within mixed populations or infection models. Beyond mechanistic insights, these tools have translational potential: TreS-specific inhibitors, such as validamycin A, 6-azido-6-deoxy-d-trehalose (6-TreAz), and 2-amino-2-deoxy-d-trehalose (2-TreNH2), have shown promise in suppressing the trehalose-catalytic shift, reducing persister formation, and resensitizing Mtb to existing antibiotics (100, 102, 106). Concurrently, RMR-tre labeling could support diagnostic and screening platforms to monitor treatment response or identify compounds that target metabolic heterogeneity.
Together, the TreS-mediated trehalose-catalytic shift and the RMR-tre labeling assay illuminate a metabolic logic underpinning Mtb’s persistence and drug resistance. These findings indicate that antibiotic tolerance and resistance arise not solely from genetic mutations but from dynamic, reversible shifts in carbon flux and redox metabolism. Targeting this metabolic plasticity, in addition to static genetic determinants, offers a promising avenue for eradicating Mtb persister populations and mitigating the evolution of multidrug-resistant TB.
B. Phosphoenolpyruvate depletion: Off/Disabled Toggle Switch (Fig. 3)
Phosphoenolpyruvate (PEP) is a pivotal metabolite at the crossroads of glycolysis, gluconeogenesis, and the tricarboxylic acid (TCA) cycle, linking energy metabolism with biosynthesis and redox balance. In bacteria, PEP donates phosphoryl groups for glycolysis and for sugar transport via the phosphotransferase system, underscoring its essential role in cellular energy balance and growth (107, 108, 109). Under hypoxia and nutrient starvation, Mtb enters a well-characterized non-replicating state, marked by decreased ATP levels and remodeling of CCM. Targeted metabolomics analyses reveal that PEP levels decline significantly during the transition to and maintenance of the non-replicating state (110, 111). Depletion of PEP closely correlates with growth kinetics and antibiotic tolerance to front-line TB antibiotics such as INH and RIF. Notably, supplementation of non-replicating Mtb cultures with exogenous PEP under in vitro hypoxia partially restores metabolic fluxes and resensitizes bacteria to INH and RIF, though the effect is incomplete. These findings indicate that PEP functions as a metabolic node that influences – but does not solely govern - the tolerant state. Nonetheless, these findings establish a causal link between PEP depletion and drug tolerance, providing a biochemical explanation for how non-replicating Mtb bacilli endure prolonged antibiotic exposure (110).
Mechanistically, PEP depletion impairs energy-generating pathways and disrupts multiple downstream anabolic processes (Fig. 3). As a high-energy intermediate in glycolysis, PEP contributes to ATP production via substrate-level phosphorylation and serves as a precursor for downstream pathways, including the TCA cycle, nascent peptidoglycan biosynthesis, and the shikimate pathway (110). Depletion likely reduces pyruvate availability, thereby constraining acetyl-CoA and TCA cycle intermediates required for respiration and maintenance of the proton motive force. The metabolic bottleneck also limits NADH turnover, altering redox balance in a manner that diminishes ROS-mediated killing typically induced by bactericidal antibiotics. In other words, low-PEP conditions shield the cells from antibiotic-induced oxidative stress by reducing metabolic activity and ROS generation - a hallmark of bacterial persistence (110).
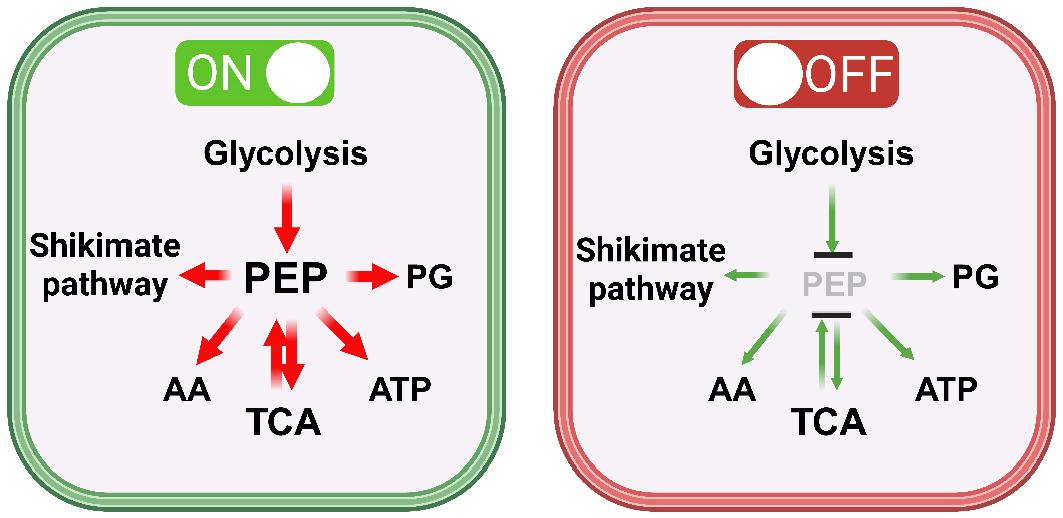
Fig. 3
Phosphoenolpyruvate (PEP) depletion as a metabolic toggle in Mtb. Schematic model illustrating how PEP availability controls flux through central carbon metabolism. Left, “ON” (PEP-replete): high PEP supports active glycolysis, carbon flux into peptidoglycan (PG) biosynthesis, the shikimate pathway, amino acid (AA) synthesis, the tricarboxylic acid (TCA) cycle, and ATP production. Right, “OFF” (PEP-depleted): under hypoxia and nutrient starvation, PEP levels fall, attenuating glycolytic flux and limiting carbon supply to PG, the shikimate pathway, AA synthesis, the TCA cycle, thereby lowering ATP output and overall metabolic activity. Created with BioRender.com.
Studies linking metabolic state to drug-resistant mutational dynamics show that PEP supplementation can increase antibiotic susceptibility and reduce the frequency of resistance-conferring mutations (110). This finding suggests that PEP-depleted cells exhibit antibiotic tolerance at the cost of metabolic constraint. This trade-off may promote the eventual development of genetic drug resistance through imbalanced DNA repair or stress-induced mutagenesis within subpopulations that survive under otherwise lethal antibiotic pressure. Thus, PEP metabolism may act as a metabolic checkpoint, functioning as a phenotypic toggle to determines whether a bacterium remains transiently tolerant or progresses toward stable, heritable resistance.
Importantly, these findings fit within a broader framework in which CCM governs Mtb’s phenotypic fate under stress. Previous studies have highlighted enzymes such as isocitrate lyase and malate dehydrogenase as critical for persistence (81, 112, 113). The current observations position PEP as an upstream metabolite that influences both growth arrest and antibiotic tolerance. The notion of a single metabolite acting as a switch between active growth and persistence underscores the plasticity of Mtb’s metabolic network and suggests new avenues aimed at destabilizing the persistence switch.
C. Succinate accumulation and secretion: Metabolic battery (Fig. 4B)
Mtb encounters hypoxic stress within granulomas, where oxygen availability is severely restricted (114, 115, 116, 117). Under these conditions, oxidative phosphorylation becomes inefficient, forcing the pathogen to remodel the TCA cycle to readjust redox balance state and ATP production. Previous studies provided a detailed analysis of this adaptive process, showing that succinate metabolism is markedly altered during adaptation to hypoxic environments (35). Using isotopic labeling and metabolomics, they demonstrated that Mtbshifts to a bifurcated TCA cycle and activates glyoxylate shunt activity that sustains the production and secretion of succinate as an electron sink.
Under normoxic conditions, succinate dehydrogenase (SDH) oxidizes succinate to fumarate, transferring electrons to the electron transport chain (ETC). However, under hypoxia, Mtbreverses this flow, reducing fumarate to succinate via fumarate reductase to regenerate oxidized cofactors (e.g., NAD⁺) required for glycolysis and anaplerotic reactions (Fig. 4) (35, 118). This metabolic remodeling enables Mtb to maintain redox homeostasis even as the ETC is downregulated. Notably, the accumulation and secretion of succinate serve as a metabolic adaptation to sustain the proton motive force (PMF), ATP synthesis, and membrane potential, thereby supporting survival during extended hypoxia. Experimental inhibition - genetic or pharmacological - of enzymes involved in succinate turnover (including SDH and malate dehydrogenase, MDH), which are largely dispensable under normoxia, markedly impairs survival as non-replicating Mtb under hypoxia exposure. This conditional essentiality highlights how flux through the succinate node becomes a metabolic lifeline during latent TB infection. Thus, succinate metabolism acts as a redox-balancing mechanism that supports anaerobic ATP generation and maintains cell viability under stress.
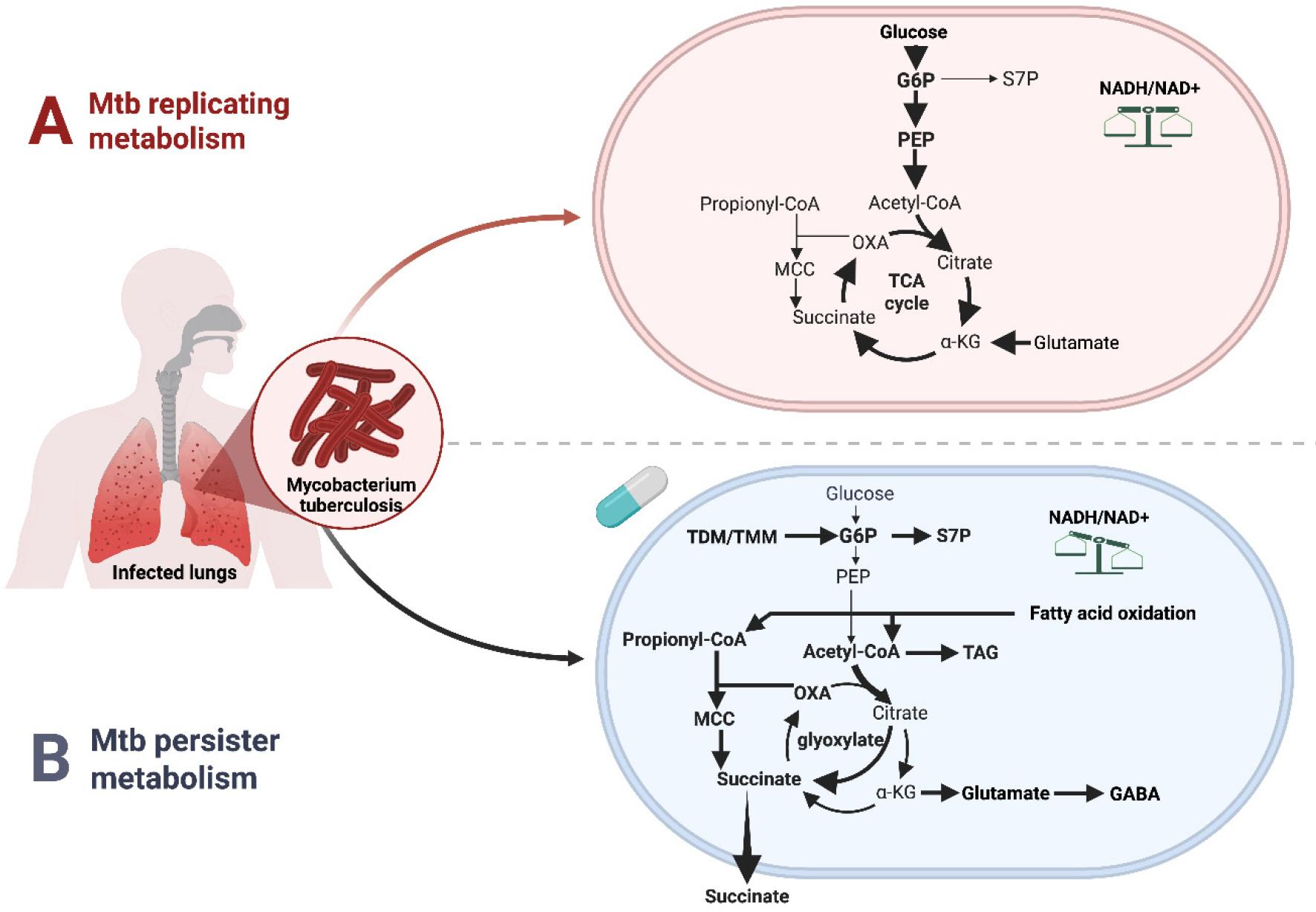
Fig. 4
Remodeling of TCA cycle and MCC in replicating versus persister Mtb. A, Replicating metabolism. Actively growing Mtb catabolizes carbohydrates through glycolysis and a tricarboxylic acid (TCA) cycle. Carbon flux through CCM while balanced NADH/NAD⁺ turnover supports oxidative phosphorylation and rapid growth.B, Persister metabolism. Under hypoxic, nutrient-limited conditions, and antibiotic exposures, Mtb shifts to a persister state in which central carbon metabolism is rewired. Propionyl-CoA produced from β-oxidation of long chain fatty acids is processed through the MCC, linking fatty acid catabolism to the TCA cycle while risking accumulation of toxic MCC intermediates (MCC toxicity). Under the conditions, the TCA cycle remodels, activating the glyoxylate shunt, thereby promoting succinate accumulation and secretion, which functions as a “metabolic battery” to maintain redox balance (NADH/NAD⁺), proton motive force, and ATP synthesis despite reduced electron transport chain activity. Carbon is diverted from the biosynthesis of α-ketoglutarate (αKG), which fuels to biosynthesize GABA and other nitrogen-rich metabolites, reflecting tight coordination between central carbon and nitrogen metabolism and redox balance that enable long-term survival of non-replicating persisters. G6P: Glucose 6 phosphates; S7P: Sedoheptulose 7 Phosphate; PEP: Phosphoenolpyruvate; OXA: Oxaloacetate; TAG: Triacylglycerol. Created with BioRender.com.
D. MCC accumulation and secretion: Metabolic toxicity (Fig. 4B)
Mtb exhibits remarkable metabolic flexibility that underpins its persistence in the hostile intracellular environment of the host (44, 66). Among adaptive pathways, fatty acid metabolism is particularly critical, as the bacillus largely utilizes host-derived long-chain fatty acids and cholesterol as major carbon and energy sources during infection (119, 120, 121, 122). The catabolic degradation of these lipids via β-oxidation generates substantial amounts of short-chain fatty acids such as acetyl-CoA and propionyl-CoA, which must be efficiently assimilated to maintain carbon flux and redox balance under nutrient limitation and hypoxia (110, 120).
The MCC serves as a central route for detoxification and assimilation of propionyl-CoA, a potentially toxic three-carbon intermediate derived from β-oxidation of odd-chain fatty acids and cholesterol catabolism (123). The role of MCC in Mtbmetabolism has been explored by constructing prpC/prpD deletion strains that cannot grow on propionate in vitro or ex vivo. The MCC is also associated with antibiotic tolerance in Mtb, with propionyl-CoA and other MCC intermediates linked to increased redox stress and tolerance to multiple antibiotics, particularly INH and RIF (124).
In the MCC, propionyl-CoA condenses with oxaloacetate to form 2-methylcitrate, which is subsequently converted through a series of enzymatic steps to succinate and pyruvate. This process prevents propionyl-CoA accumulation and replenishes TCA cycle intermediates, linking long-chain fatty acid degradation to the CCM. Under hypoxic or nutrient-starved states, MCC flux is tightly regulated (125, 126, 127). Accumulation of propionyl-CoA, 2-methylcitrate, 2-methylisocitrate, or related intermediates perturbs cellular redox balance, membrane potential, and intracellular pH, compromising bacterial viability - a phenomenon termed MCC toxicity (Fig. 4) (126, 127). To mitigate this toxicity, non-replicating Mtb must coordinate MCC activity with other anaplerotic and redox-balancing pathways, including the glyoxylate shunt, the methylmalonyl-CoA pathway, succinate secretion, and central nitrogen metabolism (35, 125, 126, 127). Notably, succinate and related MCC intermediates may be secreted as an overflow mechanism to maintain intracellular redox homeostasis and membrane potential during non-replicating latent states (35). Collectively, the MCC represents a pivotal metabolic node that integrates lipid catabolism, redox regulation, and survival under stress in Mtb.
CONCLUSION
The non-replicating latent physiology of Mtbrests on remarkable metabolic plasticity that enables survival in the nutrient-starved, hypoxic, and immune-pressured microenvironments within granulomas (44, 81, 113). Rather than a single biochemical adaptation, latency results from multifaceted reprogramming of metabolic pathways, including cell-wall glycolipid reorganization, CCM and central nitrogen metabolism remodeling, and long-chain fatty acid degradation. Together, these adjustments sustain redox balance and energy homeostasis while minimizing metabolic toxicity (Fig. 2) (127, 128). One defining feature of this plasticity is the trehalose-catalytic shift (Fig. 2), whereby trehalose-mediated carbon fluxes are redirected away from the biosynthesis of virulence-associated cell-wall glycolipids (TDM/TMM) toward catabolic generation of G6P, the first glycolysis intermediate. This transition supplies endogenous substrates for glycolysis and the PPP while downregulating fatty acid biosynthesis - a hallmark of the non-replicating latency under stress. In parallel, depletion of PEP, the downstream glycolytic intermediate, reflects a strategic throttling of glycolytic throughput that limits ATP production and ROS, thereby conferring tolerance to bactericidal antibiotics (Fig. 3) (110).
Within host-relevant microenvironments, the metabolic landscape shifts further toward succinate accumulation and secretion, with succinate acting as a redox “battery” to maintain the PMF when oxidative phosphorylation is significantly limited (Fig. 4). This rerouting of the TCA cycle toward reductive flux via fumarate reductase or glyoxylate shunt preserves NAD⁺ regeneration and supports viability under hypoxia. Complementing this, activation of the MCC detoxifies propionyl-CoA derived from host lipid catabolism, while secretion of MCC intermediates helps prevent metabolic poisoning by excess three-carbon species (Fig. 4).
Collectively, these interlocking adaptations constitute a systems-level metabolic network that tunes Mtb physiology between replication, latency, and reactivation. The resulting metabolic signatures (Figs. 2, 3, 4) - trehalose and TDM depletion, reduced PEP, succinate overflow and secretion, and MCC intermediate accumulation with secretion - form a biochemical fingerprint of latent TB infection. Emerging metabolomics data suggest that these patterns could complement immunological diagnostics by distinguishing latent TB infection from active disease or by differentiating infection with drug-sensitive versus drug-resistant strains.
Viewing latency through this metabolic lens reframes it not as a passive, non-replicating state but as a dynamic equilibrium driven by adaptive biochemical circuitry. Targeting the enzymes or flux nodes underlying these metabolic reprogramming events offers promising avenues for therapies that destabilize latency, resensitize persisters to antibiotics, and potentially shorten TB treatment regimens. Additionally, there is growing interest in leveraging biochemical and metabolic signatures as latent TB infection and drug-resistance diagnostics, integrating host immune responses and Mtb-specific metabolic alterations. Diagnostics capable of distinguishing latent from active TB and identifying infection caused by drug-sensitive or MDR/XDR-TB would be particularly valuable for informing optimal treatment strategies.
